
NILOTINIB BIOGARAN 200 mg, gélule, boîte de 28 plaquettes prédécoupées de 1
Dernière révision : 12/09/2024
Taux de TVA : 2.1%
Prix de vente : 310,59 €
Taux remboursement SS : 100%
Base remboursement SS : 310,59 €
Laboratoire exploitant : BIOGARAN
Source :

NILOTINIB BIOGARAN est indiqué dans le traitement :
· des patients adultes et pédiatriques atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive en phase chronique nouvellement diagnostiquée ;
· des patients adultes atteints de LMC chromosome Philadelphie positive (Ph+) en phase chronique et en phase accélérée, résistants ou intolérants à un traitement antérieur incluant l'imatinib. Les données d'efficacité chez les patients ayant une LMC en crise blastique ne sont pas disponibles ;
· des patients pédiatriques atteints de LMC chromosome Philadelphie positive en phase chronique, résistants ou intolérants à un traitement antérieur incluant l'imatinib.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Aplasie médullaire
Le traitement par nilotinib est associé à une thrombopénie, une neutropénie et une anémie (de grade 3 et 4 selon la classification internationale « National Cancer Institute Common ToxicityCriteria »). Leur incidence est plus fréquente chez les patients atteints de LMC résistants ou intolérants à l'imatinib, en particulier chez les patients en phase accélérée. Il convient de pratiquer un hémogramme complet toutes les deux semaines pendant les 2 premiers mois, puis une fois par mois par la suite, ou lorsque cela est cliniquement justifié. En général, l'aplasie médullaire a été réversible et a pu être traitée par une interruption temporaire du traitement par nilotinib ou en diminuant la posologie (voir rubrique Posologie et mode d'administration).
Allongement de l'intervalle QT
Il a été constaté que le nilotinib entraînait un allongement de la repolarisation ventriculaire cardiaque, mesurée par l'intervalle QT sur l'ECG, de manière dépendante de la concentration chez les patients adultes et pédiatriques.
Au cours de l'étude de phase III menée chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée recevant 300 mg de nilotinib deux fois par jour, la variation moyenne de l'intervalle QTcF à l'état d'équilibre a été de 6 ms par rapport à l'inclusion. Aucun patient n'a présenté d'intervalle QTcF > 480 ms. Aucun épisode de torsades de pointes n'a été observé.
Au cours de l'étude de phase II menée chez des patients atteints de LMC en phase chronique et en phase accélérée résistants ou intolérants à l'imatinib, recevant 400 mg de nilotinib deux fois par jour, la variation moyenne de l'intervalle QTcF à l'état d'équilibre a été de 5 ms et 8 ms, respectivement, par rapport à l'inclusion. Un intervalle QTcF > 500 ms a été observé chez < 1 % de ces patients. Au cours des études cliniques, aucun épisode de torsades de pointes n'a été observé.
Dans une étude menée chez des volontaires sains au cours de laquelle les expositions ont été comparables à celles observées chez les patients, la variation moyenne de l'intervalle QTcF, après soustraction des valeurs obtenues avec le placebo, a été de 7 ms par rapport à l'inclusion (IC ± 4 ms). Aucun patient n'a présenté d'intervalle QTcF > 450 ms. En outre, aucun cas d'arythmies cliniquement significatives n'a été observé pendant l'essai, et notamment aucun épisode de torsades de pointes (transitoires ou persistantes).
Un allongement significatif de l'intervalle QT peut survenir en cas de prise inappropriée de nilotinib avec des inhibiteurs puissants du CYP3A4 et/ou des médicaments connus pour être susceptibles d'induire un allongement de l'intervalle QT et/ou des aliments (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). En présence d'une hypokaliémie ou d'une hypomagnésémie, cet effet peut se renforcer. Un allongement de l'intervalle QT peut exposer les patients à un risque de complications fatales.
Le nilotinib doit être utilisé avec prudence chez les patients présentant un allongement de l'intervalle QTc ou présentant un risque significatif de développer un allongement de l'intervalle QTc, tels que ceux :
· présentant un allongement congénital de l'intervalle QT ;
· présentant une maladie cardiaque non contrôlée ou significative (y compris un infarctus du myocarde récent, une insuffisance cardiaque congestive, un angor instable ou une bradycardie cliniquement significative) ;
· prenant des médicaments anti-arythmiques ou d'autres substances entraînant un allongement de l'intervalle QT.
Il est conseillé de surveiller étroitement un effet sur l'intervalle QTc et de réaliser un ECG initial avant l'instauration du traitement par nilotinib et lorsque cela est cliniquement indiqué. L'hypokaliémie ou l'hypomagnésémie doivent être corrigées avant l'administration du nilotinib, et ces paramètres doivent être régulièrement surveillés au cours du traitement.
Mort subite
Des cas peu fréquents (0,1 à 1 %) de mort subite ont été rapportés chez des patients atteints de LMC en phase chronique ou en phase accélérée résistants ou intolérants à l'imatinib et ayant des antécédents de cardiopathies ou de facteurs de risques cardiaques significatifs. Des comorbidités associées à l'hémopathie maligne sous-jacente ainsi que des traitements concomitants étaient également fréquemment présents. Les anomalies de repolarisation ventriculaire ont pu être des facteurs contributifs. Aucun cas de mort subite n'a été rapporté au cours de l'étude de phase III menée chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée.
Rétention hydrique et oedème
Des formes sévères de rétention hydrique liée au médicament telles que l'épanchement pleural, l'oedème pulmonaire et l'épanchement péricardique ont été peu fréquemment observées (0,1 à 1 %) dans une étude de phase III menée chez des patients atteints de LMC nouvellement diagnostiquée. Des événements similaires ont été observés lors du suivi post-commercialisation. Une prise de poids rapide et inattendue doit être évaluée avec attention. En cas d'apparition de signes de rétention hydrique sévère au cours du traitement par nilotinib, l'étiologie doit être recherchée et les patients traités en conséquence (voir rubrique Posologie et mode d'administration pour les instructions concernant la prise en charge des toxicités non-hématologiques).
Evénements cardiovasculaires
Des événements cardiovasculaires ont été rapportés au cours d'une étude randomisée de phase III menée chez des patients atteints de LMC nouvellement diagnostiquée et observés lors du suivi post-commercialisation. Dans cette étude clinique dont la durée d'exposition au traitement médiane était de 60,5 mois, les événements cardiovasculaires de grade 3 et 4 incluaient une artériopathie oblitérante périphérique (1,4 % et 1,1 % à 300 mg et 400 mg de nilotinib deux fois par jour, respectivement), une cardiopathie ischémique (2,2 % et 6,1 % à 300 mg et 400 mg de nilotinib deux fois par jour, respectivement) et des accidents vasculaires cérébraux ischémiques (1,1 % et 2,2 % à 300 mg et 400 mg de nilotinib deux fois par jour, respectivement). Il convient de conseiller aux patients de consulter immédiatement un médecin en cas de signes ou symptômes d'événements cardiovasculaires. L'état cardiovasculaire des patients doit être évalué et les facteurs de risque cardiovasculaire surveillés et activement pris en charge au cours du traitement par nilotinib, selon les recommandations standard.
Un traitement approprié doit être prescrit pour contrôler les facteurs de risque cardiovasculaire (voir rubrique Posologie et mode d'administration pour les instructions concernant la prise en charge des toxicités non-hématologiques).
Réactivation de l'hépatite B
Des cas de réactivation de l'hépatite B ont été rapportés chez des patients porteurs chroniques du virus et traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante entraînant une transplantation hépatique ou dont l'issue a été fatale.
Les patients doivent subir un test de dépistage de l'infection par le VHB avant l'instauration d'un traitement par nilotinib. Un médecin spécialisé en hépatologie doit être consulté avant l'instauration du traitement chez les patients porteurs de marqueurs sérologiques positifs (y compris ceux ayant une hépatite B active) et chez les patients dont la sérologie devient positive au cours du traitement. Les patients porteurs du VHB doivent faire l'objet d'une surveillance étroite tout au long du traitement par nilotinib et plusieurs mois après la fin du traitement (voir rubrique Effets indésirables).
Surveillance particulière des patients adultes atteints de LMC Ph+ en phase chronique ayant obtenu le maintien d'une réponse moléculaire profonde
Eligibilité pour l'arrêt du traitement
Pour les patients éligibles chez qui l'expression des transcrits BCR-ABL types e13a2/b2a2 ou e14a2/b3a2 est confirmée, l'arrêt du traitement peut être envisagé. Les patients doivent présenter des transcrits BCR-ABL caractéristiques pour permettre la quantification BCR-ABL, l'évaluation de la profondeur de la réponse moléculaire et la détermination d'une éventuelle perte de rémission moléculaire après l'arrêt du traitement par nilotinib.
Surveillance des patients ayant arrêté le traitement
Une surveillance fréquente des taux de transcrits BCR-ABL doit être effectuée chez les patients éligibles à l'arrêt du traitement, à l'aide d'un test de diagnostic quantitatif validé, afin de mesurer les niveaux de réponse moléculaire avec une sensibilité d'au moins RM4.5 (BCR-ABL/ABL ≤ 0,0032 %EI). Les taux de transcrits BCR-ABL doivent être évalués avant et pendant l'arrêt du traitement (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques).
La perte de réponse moléculaire majeure (RMM = BCR-ABL/ABL ≤ 0,1 % EI) chez les patients atteints de LMC ayant reçu le nilotinib en première ou deuxième ligne de traitement ou la perte confirmée de RM4 (deux mesures consécutives séparées d'au moins 4 semaines indiquant la perte de RM4 [RM4 = BCR-ABL/ABL ≤ 0,01 % EI]) chez les patients atteints de LMC ayant reçu le nilotinib en deuxième ligne de traitement déclencheront la reprise du traitement dans les 4 semaines qui suivent la date connue de la perte de rémission. Une rechute moléculaire peut survenir durant la phase sans traitement, et les données sur les résultats à long terme ne sont pas encore disponibles. Il est par conséquent crucial d'effectuer une surveillance fréquente des taux de transcrits BCR-ABL et une formule sanguine complète avec numération différentielle afin de détecter une éventuelle perte de rémission (voir rubrique Posologie et mode d'administration). Pour les patients qui n'ont pas récupéré une RMM après trois mois de ré-instauration du traitement, le test de mutation du domaine kinase BRC-ABL doit être effectué.
Examens de laboratoire et surveillance
Lipémie
Au cours d'une étude de phase III menée chez des patients atteints de LMC nouvellement diagnostiquée, 1,1 % des patients traités par 400 mg de nilotinib deux fois par jour ont présenté une augmentation du cholestérol total de grade 3-4 ; toutefois, aucune augmentation de grade 3-4 n'a été observée dans le groupe recevant 300 mg deux fois par jour (voir rubrique Effets indésirables). Il est recommandé de déterminer les profils lipidiques des patients avant d'instaurer un traitement par nilotinib, de les évaluer aux Mois 3et 6après l'instauration du traitement et au minimum chaque année pendant le traitement chronique (voir rubrique Posologie et mode d'administration). Si un traitement par inhibiteur de la HMG-CoA réductase (un agent hypolipémiant) est nécessaire, il convient de se référer à la rubrique Interactions avec d'autres médicaments et autres formes d'interactions avant d'instaurer le traitement car certains inhibiteurs de la HMG-CoA réductase sont également métabolisés par la voie du CYP3A4.
Glycémie
Au cours d'une étude de phase III menée chez des patients atteints de LMC nouvellement diagnostiquée, 6,9 % et 7,2 % des patients traités respectivement par 400 mg de nilotinib et 300 mg de nilotinib deux fois par jour, ont présenté une augmentation de la glycémie de grade 3-4. Il est recommandé d'évaluer la glycémie avant d'instaurer un traitement par nilotinib et de la surveiller au cours du traitement, si cela est cliniquement indiqué (voir rubrique Posologie et mode d'administration). Si les résultats justifient un traitement, les médecins doivent suivre les recommandations thérapeutiques et de bonne pratique locale.
Interactions avec d'autres médicaments
L'administration de nilotinib avec des inhibiteurs puissants du CYP3A4 (incluant, sans s'y limiter, kétoconazole, itraconazole, voriconazole, clarithromycine, télithromycine, ritonavir) doit être évitée. Si l'administration de l'un de ces agents s'avère nécessaire, il est recommandé, si possible, d'interrompre le traitement par nilotinib (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Si l'interruption temporaire du traitement n'est pas possible, une surveillance étroite du patient est recommandée, afin d'identifier un éventuel allongement de l'intervalle QT (voir rubriques Posologie et mode d'administration, Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques).
L'utilisation concomitante de nilotinib et d'inducteurs puissants du CYP3A4 (par exemple, phénytoïne, rifampicine, carbamazépine, phénobarbital et millepertuis) réduit vraisemblablement l'exposition au nilotinib de manière cliniquement significative. Par conséquent, il convient d'opter pour une administration concomitante de médicaments ayant un faible potentiel d'induction du CYP3A4 chez les patients recevant du nilotinib (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Effets des aliments
La biodisponibilité du nilotinib est augmentée par la prise alimentaire. NILOTINIB BIOGARAN ne doit pas être pris avec des aliments (voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions) et il doit être pris 2 heures après un repas. Le patient ne doit consommer aucun aliment pendant une heure au moins après la prise du médicament. Le jus de pamplemousse et les autres aliments connus pour inhiber le CYP3A4 doivent être évités.
Pour les patients présentant des difficultés à avaler, y compris les patients pédiatriques qui ne sont pas en mesure d'avaler des gélules, d'autres médicaments à base de nilotinib doivent être utilisés à la place de NILOTINIB BIOGARAN.
Insuffisance hépatique
L'insuffisance hépatique a un effet modéré sur la pharmacocinétique du nilotinib. L'administration unique d'une dose de 200 mg de nilotinib a entraîné des augmentations de l'ASC de 35 %, 35 % et 19 % respectivement, chez des sujets présentant une insuffisance hépatique légère, modérée et sévère par rapport aux sujets du groupe témoin présentant une fonction hépatique normale. La Cmax prédictive à l'état d'équilibre du nilotinib était augmentée de 29 %, 18 % et 22 %, respectivement. Au cours des études cliniques, les patients présentant des taux d'alanine aminotransférase (ALAT) et/ou d'aspartate aminotransférase (ASAT) > 2,5 fois la limite supérieure de la normale (ou > 5 × LSN, si l'augmentation était liée à la maladie) et/ou des taux de bilirubine totale > 1,5 fois la limite supérieure de la normale, n'ont pas été inclus. Le métabolisme du nilotinib est essentiellement hépatique. Les patients présentant une insuffisance hépatique peuvent donc avoir une exposition augmentée au nilotinib et doivent être traités avec prudence (voir rubrique Posologie et mode d'administration).
Taux sériques de lipase
Une élévation des taux sériques de lipase a été observée. La prudence est de mise chez les patients ayant des antécédents de pancréatite. En cas d'élévation des taux de lipases associés à des symptômes abdominaux, le traitement par nilotinib doit être interrompu et des mesures appropriées pour établir le diagnostic et exclure une pancréatite doivent être envisagées.
Gastrectomie totale
La biodisponibilité du nilotinib peut être diminuée chez les patients ayant une gastrectomie totale (voir rubrique Propriétés pharmacocinétiques). Un suivi plus fréquent de ces patients doit être envisagé.
Syndrome de lyse tumorale
En raison de la survenue possible de syndrome de lyse tumorale (SLT), il est recommandé de corriger toute déshydratation cliniquement significative et de traiter l'hyperuricémie avant l'instauration du traitement par nilotinib (voir rubrique Effets indésirables).
Population pédiatrique
Des anomalies biologiques de type élévations transitoires légères à modérées des aminotransférases et de la bilirubine totale ont été observées chez l'enfant à une fréquence plus élevée que chez l'adulte, indiquant un risque plus élevé d'hépatotoxicité dans la population pédiatrique (voir rubrique Effets indésirables). La fonction hépatique (taux de bilirubine et de transaminases hépatiques) doit être surveillée tous les mois ou lorsque cela est cliniquement indiqué. Les élévations de la bilirubine et des transaminases hépatiques doivent être traitées par une interruption temporaire du nilotinib, une réduction de la dose et/ou un arrêt du nilotinib (voir rubrique Posologie et mode d'administration). Au cours d'une étude menée chez des patients pédiatriques atteints de LMC, un retard de croissance a été mis en évidence chez des patients traités par nilotinib (voir rubrique Effets indésirables). Une surveillance étroite de la croissance chez les patients pédiatriques sous traitement par nilotinib est recommandée.
Lactose
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Le profil de sécurité repose sur les données combinées de 3 422 patients traités par nilotinib au cours de 13 études cliniques dans les indications approuvées : patients adultes et pédiatriques atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive en phase chronique nouvellement diagnostiquée (5 études cliniques portant sur 2 414 patients), patients adultes atteints de LMC chromosome Philadelphie positive en phase chronique et en phase accélérée, résistants ou intolérants à un traitement antérieur incluant l'imatinib (6 études cliniques portant sur 939 patients) et patients pédiatriques atteints de LMC chromosome Philadelphie positive en phase chronique, résistants ou intolérants à un traitement antérieur incluant l'imatinib (2 études cliniques portant sur 69 patients). Ces données combinées représentent 9 039,34 patient-années d'exposition.
Le profil de sécurité du nilotinib est cohérent dans toutes les indications.
Les effets indésirables les plus fréquents (incidence = 15 %) issus des données de sécurité combinées étaient : un rash (26,4 %), une infection des voies aériennes supérieures (incluant pharyngite, rhinopharyngite, rhinite) (24,8 %), des céphalées (21,9 %), une hyperbilirubinémie (incluant une bilirubine sanguine augmentée) (18,6 %), des arthralgies (15,8 %), une fatigue (15,4 %), des nausées (16,8 %), un prurit (16,7 %) et une thrombopénie (16,4 %).
Tableau listant les effets indésirables
Les effets indésirables observés dans les études cliniques et lors du suivi post-commercialisation (tableau 3) sont répertoriés par classe de systèmes d'organes (classification MedDRA) et catégorie de fréquence. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3 Effets indésirables
Infections et infestations | |
Très fréquent : | Infection des voies aériennes supérieures (incluant pharyngite, rhinopharyngite, rhinite) |
Fréquent : | Folliculite, bronchite, candidose (incluant candidose orale), pneumonie, gastro-entérite, infection des voies urinaires |
Peu fréquent : | Infection par le virus de l'herpès, abcès anal, candidose (infection à Candida), furoncle, sepsis, abcès sous-cutané, pied d'athlète |
Rare : | Réactivation de l'hépatite B |
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) | |
Peu fréquent : | Papillome cutané |
Rare : | Papillome buccal, paraprotéinémie |
Affections hématologiques et du système lymphatique | |
Très fréquent : | Anémie, thrombopénie |
Fréquent : | Leucopénie, leucocytose, neutropénie, thrombocytémie |
Peu fréquent : | Eosinophilie, neutropénie fébrile, lymphopénie, pancytopénie |
Affections du système immunitaire | |
Peu fréquent : | Hypersensibilité |
Affections endocriniennes | |
Très fréquent : | Retard de croissance |
Fréquent : | Hypothyroïdie |
Peu fréquent : | Hyperthyroïdie |
Rare : | Hyperparathyroïdie secondaire, thyroïdite |
Troubles du métabolisme et de la nutrition | |
Fréquent : | Déséquilibre électrolytique (incluant hypomagnésémie, hyperkaliémie, hypokaliémie, hyponatrémie, hypocalcémie, hypercalcémie, hyperphosphatémie), diabète, hyperglycémie, hypercholestérolémie, hyperlipidémie, hypertriglycéridémie, appétit diminué, goutte, hyperuricémie, hypophosphatémie (incluant phosphore sanguin diminué) |
Peu fréquent : | Déshydratation, appétit augmenté, dyslipidémie, hypoglycémie |
Rare : | Trouble de l'appétit, syndrome de lyse tumorale |
Affections psychiatriques | |
Fréquent : | Dépression, insomnie, anxiété |
Peu fréquent : | Amnésie, état confusionnel, désorientation |
Rare : | Dysphorie |
Affections du système nerveux | |
Très fréquent : | Céphalées |
Fréquent : | Sensations vertigineuses, hypoesthésie, paresthésie, migraine |
Peu fréquent : | Accident vasculaire cérébral, hémorragie intracrânienne/cérébrale, accident vasculaire cérébral ischémique, accident ischémique transitoire, infarctus cérébral, perte de conscience (incluant syncope), tremblements, troubles de l'attention, hyperesthésie, dysesthésie, léthargie, neuropathie périphérique, syndrome des jambes sans repos, paralysie faciale |
Rare : | Sténose de l'artère basilaire, oedème cérébral, névrite optique |
Affections oculaires | |
Fréquent : | Conjonctivite, sécheresse oculaire (incluant xérophtalmie), irritation oculaire, hyperémie (sclérale, conjonctivale, oculaire), vision trouble |
Peu fréquent : | Défauts visuels, hémorragie conjonctivale, baisse de l'acuité visuelle, oedème palpébral, blépharite, photopsie, conjonctivite allergique, diplopie, hémorragie oculaire, douleurs oculaires, prurit oculaire, gonflement oculaire, maladie de la surface oculaire, oedème périorbitaire, photophobie |
Rare : | Choriorétinopathie, oedème papillaire |
Affections de l'oreille et du labyrinthe | |
Fréquent : | Vertige, douleur auriculaire, acouphènes |
Peu fréquent : | Troubles de l'audition (hypoacousie) |
Affections cardiaques | |
Fréquent : | Angine de poitrine, arythmie (incluant bloc auriculo-ventriculaire, flutter cardiaque, extrasystoles ventriculaires, tachycardie, fibrillation auriculaire, bradycardie), palpitations, allongement de l'intervalle QT à l'électrocardiogramme, maladie de l'artère coronaire |
Peu fréquent : | Infarctus du myocarde, souffle cardiaque, épanchement péricardique, insuffisance cardiaque, dysfonctionnement diastolique, bloc de branche gauche, péricardite |
Rare : | Cyanose, fraction d'éjection diminuée |
Fréquence indéterminée : | Dysfonction ventriculaire |
Affections vasculaires | |
Fréquent : | Hypertension, bouffées congestives, artériopathie oblitérante périphérique |
Peu fréquent : | Crise hypertensive, claudication intermittente, sténose artérielle périphérique, hématome, artériosclérose, hypotension, thrombose |
Rare : | Choc hémorragique |
Affections respiratoires, thoraciques et médiastinales | |
Très fréquent : | Toux |
Fréquent : | Dyspnée, dyspnée d'effort, épistaxis, douleurs oropharyngées |
Peu fréquent : | Œdème pulmonaire, épanchement pleural, pneumopathie interstitielle, douleur pleurale, pleurésie, irritation de la gorge, dysphonie, hypertension pulmonaire, sibilances |
Rare : | Douleur pharyngolaryngée |
Affections gastro-intestinales | |
Très fréquent : | Nausées, douleurs abdominales hautes, constipation, diarrhée, vomissements |
Fréquent : | Pancréatite, gêne abdominale, distension abdominale, flatulences, douleurs abdominales, dyspepsie, gastrite, reflux gastro-oesophagien, hémorroïdes, stomatite |
Peu fréquent : | Hémorragie gastro-intestinale, méléna, ulcération buccale, douleurs oesophagiennes, bouche sèche, sensibilité dentaire (hyperesthésie dentaire), dysgueusie, entérocolite, ulcère gastrique, gingivite, hernie hiatale, hémorragie rectale |
Rare : | Perforation d'un ulcère gastro-intestinal, hématémèse, ulcère de l'oesophage, oesophagite ulcéreuse, hémorragie rétropéritonéale, subiléus |
Affections hépatobiliaires | |
Très fréquent : | Hyperbilirubinémie (incluant augmentation de la bilirubinémie) |
Fréquent : | Fonction hépatique anormale |
Peu fréquent : | Hépatotoxicité, hépatite toxique, ictère, cholestase, hépatomégalie |
Affections de la peau et du tissu sous-cutané | |
Très fréquent : | Rash, prurit, alopécie |
Fréquent : | Sueurs nocturnes, eczéma, urticaire, hyperhidrose, contusion, acné, dermatite (incluant dermatites allergiques, exfoliatives et acnéiformes), sécheresse cutanée, érythème |
Peu fréquent : | Rash avec exfoliation, éruption d'origine médicamenteuse, douleurs cutanées, ecchymoses, gonflement du visage, éruption bulleuse, kystes épidermoïdes, érythème noueux, hyperkératose, pétéchies, photosensibilité, psoriasis, décoloration cutanée, desquamation cutanée, hyperpigmentation cutanée, hypertrophie cutanée, ulcère cutané |
Rare : | Erythème polymorphe, érythrodysesthésie palmo-plantaire, hyperplasie sébacée, atrophie cutanée |
Affections musculosquelettiques et du tissu conjonctif | |
Très fréquent : | Myalgies, arthralgies, dorsalgies, douleurs dans les extrémités |
Fréquent : | Douleurs musculosquelettiques au niveau du thorax, douleurs musculosquelettiques, cervicalgies, faiblesse musculaire, spasmes musculaires, douleurs osseuses |
Peu fréquent : | Raideur musculosquelettique, tuméfaction articulaire, arthrite, douleurs costales |
Affections du rein et des voies urinaires | |
Fréquent : | Pollakiurie, dysurie |
Peu fréquent : | Impériosité mictionnelle, nycturie, chromaturie, hématurie, insuffisance rénale, incontinence urinaire |
Affections des organes de reproduction et du sein | |
Fréquent : | Dysérection, ménorragie |
Peu fréquent : | Douleurs mammaires, gynécomastie, gonflement du mamelon |
Rare : | Induration mammaire |
Troubles généraux et anomalies au site d'administration | |
Très fréquent : | Fatigue, fièvre |
Fréquent : | Douleurs thoraciques (incluant douleurs thoraciques non cardiaques), douleurs, gêne thoracique, malaise, asthénie et oedème périphérique, frissons, syndrome grippal |
Peu fréquent : | Œdème de la face, oedème gravitationnel, sensations de modification de la température corporelle (sensations de chaleur, sensations de froid), oedème localisé |
Rare : | Mort subite |
Investigations | |
Très fréquent : | Alanine aminotransférase augmentée, lipase augmentée |
Fréquent : | Hémoglobine diminuée, amylase sanguine augmentée, aspartate aminotransférase augmentée, phosphatase alcaline sanguine augmentée, gamma-glutamyltransférase augmentée, créatinine phosphokinase sanguine augmentée, poids diminué, poids augmenté, taux élevé de créatinine, cholestérol total augmenté |
Peu fréquent : | Lactate déshydrogénase sanguine augmentée, urée sanguine augmentée, bilirubine libre sanguine augmentée, parathormone sanguine augmentée, triglycérides sanguins augmentés, diminution des globulines, lipoprotéines (incluant LDL et HDL) augmentées, troponine augmentée |
Rare : | Glucose sanguin diminué, insuline sanguine diminuée, insuline sanguine augmentée, peptide C-insuline diminué |
Remarque : les effets indésirables n'ont pas tous été observés au cours des études pédiatriques.
Description de certains effets indésirables
Mort subite
Des cas peu fréquents (0,1 à 1 %) de mort subite ont été rapportés au cours d'études cliniques portant sur le nilotinib et/ou dans le cadre de programmes d'usage compassionnel chez des patients atteints de LMC en phase chronique ou en phase accélérée résistants ou intolérants à l'imatinib ayant des antécédents de cardiopathie ou des facteurs de risque cardiaques significatifs associés (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactivation de l'hépatite B
Des cas de réactivation de l'hépatite B ont été rapportés en association avec des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante entraînant une transplantation hépatique ou dont l'issue a été fatale (voir rubrique Mises en garde spéciales et précautions d'emploi).
Population pédiatrique
La sécurité du nilotinib chez les patients pédiatriques (âgés de 2 à < 18 ans) atteints de LMC chromosome Philadelphie positive en phase chronique (n = 58) a été étudiée au cours d'une étude principale couvrant une période de 60 mois (voir rubrique Propriétés pharmacodynamiques). Chez les patients pédiatriques, la fréquence, le type et la sévérité des effets indésirables observés étaient généralement comparables à ceux observés chez les adultes, à l'exception d'une hyperbilirubinémie/élévation de la bilirubine sanguine (grade 3/4 : 10,3 %) et une élévation des transaminases (ASAT de grade 3/4 ; 1,7 %, ALAT de grade 3/4 ; 12,1 %) qui ont été rapportées à une fréquence plus élevée que chez les patients adultes. Les taux de bilirubine et de transaminases hépatiques doivent être contrôlés au cours du traitement (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Retard de croissance dans la population pédiatrique
Au cours d'une étude effectuée dans la population pédiatrique atteinte de LMC, avec une exposition médiane de 51,9 mois chez des patients nouvellement diagnostiqués et de 59,9 mois chez des patients présentant une LMC Ph+ en phase chronique résistants à l'imatinib/au dasatinib ou intolérants à l'imatinib, une décélération de la croissance (dépassant au moins deux lignes de percentile principales par rapport à l'inclusion) a été observée chez huit patients : cinq patients (8,6 %) ont dépassé deux lignes de percentile principales par rapport à l'inclusion et trois patients (5,2 %) ont dépassé trois lignes de percentile principales par rapport à l'inclusion. Des événements liés à un retard de croissance ont été rapportés chez 3 patients (5,2 %). Une surveillance étroite de la croissance chez les patients pédiatriques traités par nilotinib est recommandée (voir rubrique Mises en garde spéciales et précautions d'emploi).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
SURVEILLANCE AVANT l'instauration
du traitement par nilotinib :
- Réaliser un ECG initial.
- Corriger toute
déshydratation cliniquement significative et traiter l'hyperuricémie.
-
Corriger l'hypokaliémie ou l'hypomagnésémie.
- Faire un bilan lipidique.
- Faire un dépistage de
l'infection par le VHB. Un médecin spécialisé en hépatologie doit être consulté
avant instauration du traitement chez les patients porteurs de marqueurs
sérologiques positifs (y compris ceux ayant une hépatite B active) et chez les
patients dont la sérologie devient positive en cours du traitement. Les patients
porteurs du VHB doivent être étroitement suivis tout au long du traitement par
le nilotinib et plusieurs mois après la fin du traitement.
- Evaluer la
glycémie.
SURVEILLANCE PENDANT le traitement :
- Etat cardiovasculaire.
- ECG lorsque cela est
cliniquement justifié.
- Hémogramme complet toutes les deux semaines pendant
les deux premiers mois, puis une fois par mois par la suite, ou lorsque cela est
cliniquement justifié.
- Kaliémie et magnésémie.
- Bilan lipidique évalué
aux 3ème mois et 6ème mois et au minimum chaque année pendant le traitement
chronique.
- Glycémie.
- Fonction hépatique dans la population pédiatrique : bilirubinémie et
taux de transaminases une fois par mois ou lorsque cela est cliniquement
justifié.
- Lipasémie une fois par mois ou lorsque cela est cliniquement
justifié.
SURVEILLANCE APRES arrêt du traitement : taux de transcrit BCR-ABL
et formule sanguine complète avec numération différentielle tous les mois
pendant un an, puis toutes les 6 semaines la deuxième année, et ensuite toutes
les 12 semaines. La surveillance des taux de transcrit BCR-ABL doit être
effectuée avec un test de diagnostic quantitatif validé pour mesurer les taux de
réponse moléculaire sur l'échelle internationale (EI) avec une sensibilité d'au
moins RM4.5 (BCR-ABL/ABL ≤0,0032% EI).
PREVENIR IMMEDIATEMENT UN MEDECIN
en cas de signes de :
- douleurs
musculosquelettiques : douleurs dans les articulations et les muscles ;
-
troubles cardiaques : douleur ou gêne au niveau du thorax, pression artérielle
élevée ou basse, rythme cardiaque irrégulier, palpitations, évanouissement,
décoloration bleue des lèvres, de la langue ou de la peau ;
- obstruction
artérielle : douleur, gêne, faiblesse ou crampe musculaire dans les jambes,
ulcères sur les jambes ou les bras qui cicatrisent mal ou pas du tout,
modification notable de la couleur ou de la température de la jambe, du bras,
des orteils ou des doigts affectés ;
- hypoactivité de la glande thyroïde :
prise de poids, chute de cheveux, faiblesse musculaire, sensation de froid ;
-
hyperactivité de la glande thyroïde : battements de coeur rapides, yeux gonflés,
perte de poids, gonflement sur l'avant du cou ;
- troubles des reins ou des
voies urinaires : soif, peau sèche, irritabilité, urines foncées, diminution du
débit urinaire, difficulté et douleur lors de l'émission d'urine, sensation
exagérée d'envie d'uriner, présence de sang dans les urines, coloration anormale
des urines ;
- taux sanguins élevés de sucre : soif excessive, débit urinaire
élevé, augmentation de l'appétit avec perte de poids, fatigue ;
- vertiges :
sensations vertigineuses ou sensation de tournoiement ;
- pancréatite :
douleurs abdominales hautes (moyennes ou gauches) sévères.
- troubles cutanés
: vésicules (« ampoules ») rouges et douloureuses, douleurs cutanées, rougeur
cutanée, desquamation ou cloques ;
- rétention d'eau : prise de poids rapide,
gonflement des mains, des chevilles, des pieds ou du visage ;
- migraine :
maux de tête sévères s'accompagnant souvent de nausées, de vomissements et d'une
sensibilité à la lumière.
- troubles sanguins : fièvre, hématomes (« bleus »)
apparaissant facilement ou saignement inexpliqué, infections sévères ou
fréquentes, faiblesse inexpliquée ;
- caillots à l'intérieur d'une veine :
gonflement et douleur dans une région du corps ;
- troubles du système nerveux
: faiblesse ou paralysie des membres ou du visage, difficultés d'élocution, maux
de tête sévères, visualisation, sensation ou audition de choses qui n'existent
pas, changements de la vue, perte de connaissance, confusion, désorientation,
tremblements, sensation de picotements, douleurs ou engourdissement dans les
doigts et les orteils ;
- troubles pulmonaires : difficultés à respirer ou
douleurs respiratoires, toux, respiration sifflante avec ou sans fièvre,
gonflement des pieds ou des jambes ;
- troubles gastro-intestinaux : douleurs abdominales, nausées, vomissements de sang, selles noires ou sanglantes, constipation, brûlures d'estomac, reflux d'acide dans l'estomac, abdomen gonflé ;
- troubles du foie : jaunissement de la peau et des yeux, nausées, perte d'appétit, urines foncées ;
- signes de troubles oculaires : troubles visuels incluant vision trouble, vision double ou perception de flashs de lumière, diminution de l'acuité visuelle ou perte de vision, saignement dans l'oeil, sensibilité accrue des yeux à la lumière, douleur, rougeur, démangeaisons ou irritation oculaires, sécheresse oculaire, gonflement ou démangeaisons au niveau des paupières ;
- signes de
déséquilibre des électrolytes : nausées, essoufflement, pouls irrégulier, urine
trouble, fatigue et/ou gêne articulaire associée à des résultats anormaux des
analyses de sang.
NE PAS PRENDRE de
pamplemousse ou de jus de pamplemousse, ainsi que du millepertuis (Hypericum perforatum) pendant le
traitement.
FEMME en AGE de PROCREER : utiliser une méthode de contraception
hautement efficace pendant le traitement par nilotinib et jusqu'à deux semaines
après la fin du traitement.
PRUDENCE en cas de conduite de véhicules ou
d'utilisation de machines (sensations vertigineuses, fatigue, troubles de la
vision ou autres effets indésirables
susceptibles d'altérer l'aptitude à conduire des véhicules ou à utiliser des
machines).
Femme en capacité de procréer/Contraception
Les femmes en capacité de procréer doivent utiliser une méthode de contraception hautement efficace pendant le traitement par nilotinib et jusqu'à deux semaines après la fin du traitement.
Grossesse
Il n'existe pas ou peu de données sur l'utilisation du nilotinib chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Le nilotinib ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la femme justifie le traitement par nilotinib. En cas d'utilisation au cours de la grossesse, la patiente doit être informée des risques potentiels pour le foetus.
Si une femme traitée par nilotinib envisage une grossesse, l'arrêt du traitement pourra être envisagé sur la base des critères d'éligibilité à l'arrêt de traitement comme cela est décrit dans les rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi. Il existe peu de données sur les grossesses de patientes qui tentent d'obtenir une rémission sans traitement (RST). Si la grossesse est planifiée pendant la phase de RST, la patiente doit être informée de l'éventuelle nécessité de réinstaurer le traitement par nilotinib pendant la grossesse (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Allaitement
On ne sait pas si le nilotinib est excrété dans le lait maternel. Les données toxicologiques disponibles chez l'animal ont révélé que le nilotinib était excrété dans le lait (voir rubrique Données de sécurité préclinique). Un risque pour les nouveau-nés/nourrissons ne pouvant être exclu, les femmes ne doivent pas allaiter pendant le traitement par nilotinib et au cours des 2 semaines suivant l'administration de la dernière dose.
Fertilité
Des études effectuées chez l'animal n'ont pas révélé d'effet sur la fertilité chez les rats mâles et femelles (voir rubrique Données de sécurité préclinique).
Le nilotinib peut être administré en association avec des facteurs de croissance hématopoïétiques tels que l'érythropoïétine ou le facteur de croissance des granulocytes (G-CSF), si cela est cliniquement indiqué. Il peut être administré avec l'hydroxyurée ou l'anagrélide si cela est cliniquement indiqué.
Le nilotinib est essentiellement métabolisé dans le foie, avec le CYP3A4 considéré comme un facteur prépondérant du métabolisme oxydatif. Le nilotinib est également un substrat de la pompe à efflux de nombreux médicaments, la glycoprotéine P (P-gp). Par conséquent, les substances exerçant un effet sur le CYP3A4 et/ou la P-gp peuvent influer sur l'absorption et l'élimination du nilotinib absorbé par voie systémique.
Substances susceptibles d'augmenter les concentrations sériques de nilotinib
L'administration concomitante de nilotinib et d'imatinib (substrat et modulateur de la P-gp et du CYP3A4), exerce un faible pouvoir inhibiteur du CYP3A4 et de la P-gp. L'ASC de l'imatinib a augmenté de 18 % à 39 %, et celle du nilotinib de 18 % à 40 %. Ces variations sont vraisemblablement sans pertinence clinique.
Chez des sujets sains, l'exposition au nilotinib était multipliée par 3 en cas d'administration concomitante de kétoconazole, un inhibiteur puissant du CYP3A4. Un traitement concomitant par des inhibiteurs puissants du CYP3A4, tels que le kétoconazole, l'itraconazole, le voriconazole, le ritonavir, la clarithromycine et la télithromycine doit donc être évité (voir rubrique Mises en garde spéciales et précautions d'emploi). Une exposition augmentée au nilotinib est également attendue avec les inhibiteurs modérés du CYP3A4. L'utilisation d'autres médicaments concomitants inhibant faiblement ou n'inhibant pas le CYP3A4 doit être envisagée.
Substances susceptibles de diminuer les concentrations sériques de nilotinib
La rifampicine, un puissant inducteur du CYP3A4, diminue de 64 % la Cmax du nilotinib et réduit l'ASC du nilotinib de 80 %. La rifampicine et le nilotinib ne doivent pas être utilisés de façon concomitante.
L'administration concomitante d'autres médicaments inducteurs du CYP3A4 (par exemple, phénytoïne, carbamazépine, phénobarbital et millepertuis) diminue aussi probablement l'exposition au nilotinib de manière cliniquement significative. Chez les patients pour lesquels les inducteurs du CYP3A4 sont indiqués, il convient d'opter pour d'autres agents entraînant une induction enzymatique plus faible.
La solubilité du nilotinib dépend du pH, avec une solubilité plus faible à pH élevé. Chez des sujets sains ayant reçu de l'ésoméprazole à la dose de 40 mg une fois par jour pendant 5 jours, le pH gastrique a été augmenté de façon significative mais l'absorption de nilotinib n'a été que peu diminuée (diminution de la Cmax de 27 % et augmentation de ASC0-8 de 34 %).
Le nilotinib peut être utilisé de façon concomitante avec l'ésoméprazole ou d'autres inhibiteurs de la pompe à protons si nécessaire.
Lors d'une étude chez les sujets sains, aucun changement significatif de la pharmacocinétique du nilotinib n'a été observé lorsqu'une dose unique de 400 mg de nilotinib a été administrée 10 heures après et 2 heures avant la famotidine. Ainsi, lorsque l'utilisation concomitante d'un anti-H2 s'avère nécessaire, ce dernier peut être administré approximativement 10 heures avant ou 2 heures après la prise de nilotinib.
Dans cette même étude, l'administration d'un anti-acide (hydroxyde d'aluminium/hydroxyde de magnésium/siméticone) 2 heures avant ou après une dose unique de 400 mg de nilotinib n'a pas non plus altéré la pharmacocinétique du nilotinib. Par conséquent, si cela s'avère nécessaire, un anti-acide pourra être administré environ 2 heures avant ou 2 heures après la prise de nilotinib.
Substances dont la concentration systémique est susceptible d'être modifiée par le nilotinib
In vitro, le nilotinib est un inhibiteur relativement puissant du CYP3A4, du CYP2C8, du CYP2C9, du CYP2D6 et de l'UGT1A1, avec une valeur du Ki qui est plus basse pour le CYP2C9 (Ki = 0,13 microM).
Au cours d'une étude d'interaction médicamenteuse menée chez des volontaires sains avec une dose unique de 25 mg de warfarine, un substrat sensible du CYP2C9, une dose de 800 mg de nilotinib n'a pas entraîné de changements des paramètres pharmacocinétiques ou pharmacodynamiques de la warfarine tels que le taux de prothrombine (TP) et le rapport international normalisé (INR). On ne dispose pas de données à l'état d'équilibre. Cette étude suggère qu'une interaction médicamenteuse cliniquement significative entre le nilotinib et la warfarine est moins probable jusqu'à une dose de 25 mg de warfarine. En raison du manque de données à l'état d'équilibre, un contrôle des marqueurs pharmacodynamiques de la warfarine (INR ou TP) est recommandé après l'instauration d'un traitement par nilotinib (au moins pendant les 2 premières semaines).
Chez les patients atteints d'une LMC, le nilotinib administré à la dose de 400 mg deux fois par jour pendant 12 jours a augmenté l'exposition systémique (ASC et Cmax) au midazolam oral (un substrat du CYP3A4) d'un facteur 2,6 et 2,0, respectivement. Le nilotinib est un inhibiteur modéré du CYP3A4. Par conséquent, l'exposition systémique à d'autres médicaments essentiellement métabolisés par le CYP3A4 (par exemple, certains inhibiteurs de la HMG-CoA réductase) pourrait être augmentée en cas d'administration concomitante avec le nilotinib. Une surveillance adéquate et une adaptation posologique peuvent être nécessaires pour les médicaments qui sont des substrats du CYP3A4 et possèdent une marge thérapeutique étroite (incluant, sans s'y limiter, l'alfentanil, la ciclosporine, la dihydroergotamine, l'ergotamine, le fentanyl, le sirolimus et le tacrolimus) en cas d'administration concomitante avec le nilotinib.
L'association du nilotinib avec ces statines qui sont majoritairement éliminées par le CYP3A4 peut augmenter le risque de myopathie induite par les statines, y compris une rhabdomyolyse.
Médicaments anti-arythmiques et autres substances susceptibles d'allonger l'intervalle QT
Le nilotinib doit être utilisé avec prudence chez les patients présentant ou pouvant développer un allongement de l'intervalle QT, notamment les patients prenant des médicaments anti-arythmiques tels que l'amiodarone, le disopyramide, le procaïnamide, la quinidine ou le sotalol, ou d'autres médicaments susceptibles d'entraîner un allongement de l'intervalle QT tels que la chloroquine, l'halofantrine, la clarithromycine, l'halopéridol, la méthadone ou la moxifloxacine (voir rubrique Mises en garde spéciales et précautions d'emploi).
Interactions avec les aliments
La prise alimentaire augmente l'absorption et la biodisponibilité du nilotinib, avec pour résultat une augmentation de la concentration sérique (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques). Le jus de pamplemousse et les autres aliments connus pour inhiber le CYP3A4 doivent être évités.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
Le traitement doit être instauré par un médecin expérimenté dans le diagnostic et le traitement de la LMC.
Posologie
Le traitement doit être poursuivi aussi longtemps qu'un bénéfice clinique est observé ou jusqu'à ce qu'il entraîne une toxicité inacceptable.
En cas d'oubli d'une dose, le patient ne doit pas prendre une dose supplémentaire, mais prendre la dose suivante habituellement prescrite.
Posologie pour les patients adultes atteints de LMC chromosome Philadelphie positive
La dose recommandée est de :
· 300 mg deux fois par jour chez les patients atteints de LMC en phase chronique nouvellement diagnostiquée,
· 400 mg deux fois par jour chez les patients atteints de LMC en phase chronique et en phase accélérée, résistants ou intolérants à un traitement antérieur.
Posologie pour les patients pédiatriques atteints de LMC chromosome Philadelphie positive
La posologie chez les patients pédiatriques est individualisée et est basée sur la surface corporelle (mg/m2). La dose recommandée du nilotinib est de 230 mg/m2 deux fois par jour, arrondie aux 50 mg les plus proches (jusqu'à une dose unique maximale de 400 mg) (voir tableau 1). Différents dosages de gélules de nilotinib peuvent être combinés pour obtenir la dose souhaitée.
On ne dispose pas d'expérience sur le traitement des patients pédiatriques âgés de moins de 2 ans. Aucune donnée n'est disponible chez les patients pédiatriques âgés de moins de 10 ans nouvellement diagnostiqués et les données relatives aux patients pédiatriques âgés de moins de 6 ans résistants ou intolérants à l'imatinib sont limitées.
Tableau 1 Schéma posologique pédiatrique du nilotinib 230 mg/m2 deux fois par jour
|
Surface corporelle (SC) |
Dose en mg (deux fois par jour) |
|
Jusqu'à 0,32 m2 |
50 mg |
|
0,33-0,54 m2 |
100 mg |
|
0,55-0,76 m2 |
150 mg |
|
0,77-0,97 m2 |
200 mg |
|
0,98-1,19 m2 |
250 mg |
|
1,20-1,41 m2 |
300 mg |
|
1,42-1,63 m2 |
350 mg |
|
≥ 1,64 m2 |
400 mg |
Patients adultes atteints de LMC chromosome Philadelphie positive en phase chronique ayant été traités par nilotinib en traitement de première ligne et ayant obtenu le maintien d'une réponse moléculaire profonde (RM4.5)
L'arrêt du traitement peut être envisagé chez les patients adultes atteints de LMC chromosome Philadelphie positive (Ph+) en phase chronique éligibles, ayant été traités par nilotinib à la posologie de 300 mg deux fois par jour pendant 3 ans minimum si une réponse moléculaire profonde est maintenue pendant au moins un an juste avant l'arrêt du traitement. L'arrêt du traitement par nilotinib doit être instauré par un médecin expérimenté dans le traitement des patients atteints de LMC (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Les taux de transcrits BCR-ABL et la formule sanguine complète avec numération différentielle doivent être surveillés tous les mois pendant un an, puis toutes les 6 semaines la deuxième année, et toutes les 12 semaines par la suite, chez les patients éligibles qui arrêtent le traitement par nilotinib. La surveillance des taux de transcrits BCR-ABL doit être effectuée à l'aide d'un test de diagnostic quantitatif validé pour mesurer les taux de réponse moléculaire sur l'échelle internationale (EI) avec une sensibilité d'au moins RM4.5 (BCR-ABL/ABL ≤ 0,0032 % EI).
Chez les patients présentant une perte de RM4 (RM4 = BCR-ABL/ABL ≤ 0,01 % EI) mais ne présentant pas de perte de RMM (RMM = BCR-ABL/ABL ≤ 0,1 % EI) durant la phase sans traitement, les taux de transcrits BCR-ABL doivent être surveillés toutes les 2 semaines jusqu'à ce que les taux BCR-ABL reviennent à une valeur comprise entre RM4 et RM4.5. Les patients qui maintiennent leurs taux BCR-ABL entre la RMM et la RM4 pendant au moins 4 mesures consécutives peuvent reprendre le schéma de surveillance initial.
Les patients présentant une perte de RMM doivent reprendre le traitement dans les 4 semaines qui suivent la date connue de la perte de rémission. Le traitement par nilotinib doit être repris à la posologie de 300 mg deux fois par jour ou à une posologie réduite de 400 mg une fois par jour si le patient avait bénéficié d'une réduction de posologie avant l'arrêt du traitement. Les taux de transcrits BCR-ABL doivent être surveillés mensuellement jusqu'à ce que la RMM soit rétablie et toutes les 12 semaines par la suite, chez les patients qui reprennent le traitement par nilotinib (voir rubrique Mises en garde spéciales et précautions d'emploi).
Patients adultes atteints de LMC chromosome Philadelphie positive en phase chronique ayant obtenu le maintien d'une réponse moléculaire profonde (RM4.5) avec le nilotinib après un traitement préalable par imatinib
L'arrêt du traitement peut être envisagé chez les patients adultes atteints de LMC chromosome Philadelphie positive (Ph+) en phase chronique éligibles, ayant été traités par nilotinib pendant au moins 3 ans si une réponse moléculaire profonde est maintenue pendant au moins un an juste avant l'arrêt du traitement. L'arrêt du traitement par nilotinib doit être instauré par un médecin expérimenté dans le traitement des patients atteints de LMC (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Les taux de transcrits BCR-ABL et la formule sanguine complète avec numération différentielle doivent être surveillés tous les mois pendant un an, puis toutes les 6 semaines la deuxième année, et toutes les 12 semaines par la suite, chez les patients éligibles qui arrêtent le traitement par nilotinib. La surveillance des taux de transcrits BCR-ABL doit être effectuée à l'aide d'un test de diagnostic quantitatif validé pour mesurer les taux de réponse moléculaire sur l'échelle internationale (EI) avec une sensibilité d'au moins RM4.5 (BCR-ABL/ABL ≤ 0.0032 % EI).
Les patients présentant une perte confirmée de RM4 (RM4 = BCR-ABL/ABL ≤ 0,01 % EI) durant la phase sans traitement (deux mesures consécutives séparées d'au moins 4 semaines indiquant une perte de RM4) ou une perte de réponse moléculaire majeure (RMM = BCR-ABL/ABL ≤ 0,1 % EI) doivent reprendre le traitement dans les 4 semaines qui suivent la date connue de la perte de rémission. Le traitement par nilotinib doit être repris à la posologie de 300 mg ou 400 mg deux fois par jour. Les taux de transcrits BCR-ABL doivent être surveillés mensuellement jusqu'à ce que la réponse moléculaire majeure ou le niveau RM4 antérieurs soient rétablis et toutes les 12 semaines par la suite, chez les patients qui reprennent le traitement par nilotinib (voir rubrique Mises en garde spéciales et précautions d'emploi).
Adaptations ou modifications de la posologie
Il peut être nécessaire d'interrompre provisoirement le traitement par nilotinib et/ou de réduire la dose en cas de toxicités hématologiques (neutropénie, thrombopénie) non liées à la leucémie sous-jacente (voir tableau 2).
Tableau 2 Adaptations posologiques en cas de neutropénie et de thrombopénie
|
Patients adultes atteints de LMC en phase chronique nouvellement diagnostiquée à la posologie de 300 mg deux fois par jour et LMC en phase chronique en cas de résistance ou intolérance à l'imatinib à la posologie de 400 mg deux fois par jour |
PN* < 1,0 × 109/L et/ou plaquettes < 50 × 109/L |
1. Le traitement par nilotinib doit être interrompu et l'hémogramme doit être surveillé. 2. Le traitement doit être repris dans les 2 semaines à la dose antérieure si les PN sont > 1,0 × 109/L et/ou si les plaquettes sont > 50 × 109/L. 3. Si les valeurs de l'hémogramme restent faibles, il pourra être nécessaire de réduire la posologie à 400 mg une fois par jour. |
|
Patients adultes atteints de LMC en phase accélérée résistants ou intolérants à l'imatinib à la posologie de 400 mg deux fois par jour |
PN* < 0,5 × 109/L et/ou plaquettes < 10 × 109/L |
1. Le traitement par nilotinib doit être interrompu et l'hémogramme doit être surveillé. 2. Le traitement doit être repris dans les 2 semaines à la dose antérieure, si les PN sont > 1,0 × 109/L et/ou si les plaquettes sont > 20 × 109/L. 3. Si les valeurs de l'hémogramme restent faibles, il pourra être nécessaire de réduire la posologie à 400 mg une fois par jour. |
|
Patients pédiatriques atteints de LMC en phase chronique nouvellement diagnostiquée à la posologie de 230 mg/m2 deux fois par jour et de LMC en phase chronique résistants ou intolérants à l'imatinib à la posologie de 230 mg/m2 deux fois par jour |
PN* < 1,0 × 109/L et/ou plaquettes < 50 × 109/L |
1. Le traitement par nilotinib doit être interrompu et l'hémogramme doit être surveillé. 2. Le traitement doit être repris dans les 2 semaines à la dose antérieure, si les PN sont > 1,5 × 109/L et/ou si les plaquettes sont > 75 × 109/L. 3. Si les valeurs de l'hémogramme restent faibles, il pourra être nécessaire de réduire la posologie à 230 mg/m2 une fois par jour. 4. Si un événement survient après une réduction de la dose, envisager l'arrêt du traitement. |
* PN = polynucléaires neutrophiles
En cas de toxicité extra-hématologique modérée ou sévère cliniquement significative, le traitement doit être interrompu, et les patients doivent être surveillés et traités en conséquence. Si la dose antérieure était de 300 mg deux fois par jour chez les patients adultes atteints de LMC en phase chronique nouvellement diagnostiquée, ou de 400 mg deux fois par jour chez les patients adultes atteints de LMC en phase chronique ou accélérée résistants ou intolérants à l'imatinib, ou de 230 mg/m2 deux fois par jour chez les patients pédiatriques, le traitement pourra être repris à la dose de 400 mg une fois par jour chez les patients adultes et à la dose de 230 mg/m2 une fois par jour chez les patients pédiatriques après résolution de la toxicité. Si la dose antérieure était de 400 mg une fois par jour chez les patients adultes ou de 230 mg/m2 une fois par jour chez les patients pédiatriques, le traitement doit être arrêté. Si cela est cliniquement justifié, une nouvelle augmentation de la posologie à une dose initiale de 300 mg deux fois par jour chez les patients adultes atteints de LMC en phase chronique nouvellement diagnostiquée ou à 400 mg deux fois par jour chez les patients adultes atteints de LMC en phase chronique ou accélérée résistants ou intolérants à l'imatinib ou à 230 mg/m2 deux fois par jour chez les patients pédiatriques doit être envisagée.
Elévation des taux sériques de lipase : en cas d'élévation des taux sériques de lipase de grade 3 ou 4 chez les patients adultes, la dose doit être réduite à 400 mg une fois par jour ou le traitement interrompu. Chez les patients pédiatriques, le traitement doit être interrompu jusqu'à ce que l'événement revienne à un grade ≤ 1. Ensuite, si la dose antérieure était de 230 mg/m2 deux fois par jour, le traitement peut être repris à la dose de 230 mg/m2 une fois par jour. Si la dose antérieure était de 230 mg/m2 une fois par jour, le traitement doit être arrêté. Les taux sériques de lipase doivent être contrôlés une fois par mois ou lorsque cela est cliniquement justifié (voir rubrique Mises en garde spéciales et précautions d'emploi).
Elévation de la bilirubine et des transaminases hépatiques : en cas d'élévation de la bilirubine et des transaminases hépatiques de grade 3 ou 4 chez les patients adultes, la dose doit être réduite à 400 mg une fois par jour ou le traitement interrompu. Pour les élévations de la bilirubine de grade ≥ 2 ou les élévations des transaminases hépatiques de grade ≥ 3 chez les patients pédiatriques, le traitement doit être interrompu jusqu'à ce que les taux reviennent à un grade ≤ 1. Ensuite, si la dose antérieure était de 230 mg/m2 deux fois par jour, le traitement peut être repris à la dose de 230 mg/m2 une fois par jour. Si la dose antérieure était de 230 mg/m2 une fois par jour, et que la récupération à un grade ≤ 1 intervient sur plus de 28 jours, le traitement doit être arrêté. La bilirubinémie et les taux de transaminases hépatiques doivent être contrôlés une fois par mois ou lorsque cela est cliniquement justifié.
Populations particulières
Sujets âgés
Environ 12 % des sujets de l'étude de phase III menée chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée et 30 % environ des sujets de l'étude de phase II menée chez des patients atteints de LMC en phase chronique et en phase accélérée résistants ou intolérants à l'imatinib, étaient âgés de 65 ans et plus. Aucune différence notable n'a été observée en termes de sécurité et d'efficacité chez les patients âgés de ≥ 65 ans par rapport aux adultes âgés de 18 à 65 ans.
Insuffisance rénale
Aucune étude clinique n'a été réalisée chez les patients présentant une altération de la fonction rénale.
Etant donné que le nilotinib et ses métabolites ne sont pas excrétés par voie rénale, une diminution de la clairance corporelle totale est peu probable chez les patients présentant une insuffisance rénale.
Insuffisance hépatique
L'insuffisance hépatique a un effet modéré sur la pharmacocinétique du nilotinib. Aucune adaptation posologique n'est jugée nécessaire chez les patients présentant une insuffisance hépatique. Toutefois, la prudence est de mise chez ces patients (voir rubrique Mises en garde spéciales et précautions d'emploi).
Affections cardiaques
Au cours des études cliniques, les patients présentant une maladie cardiaque non contrôlée ou significative (par exemple, infarctus du myocarde récent, insuffisance cardiaque congestive, angor instable ou bradycardie cliniquement significative) n'ont pas été inclus. La prudence est de mise chez les patients présentant des troubles cardiaques importants (voir rubrique Mises en garde spéciales et précautions d'emploi)
Des augmentations du taux de cholestérol sérique total ont été rapportées avec un traitement par nilotinib (voir rubrique Mises en garde spéciales et précautions d'emploi). Le profil lipidique doit être déterminé avant l'instauration d'un traitement par nilotinib, évalué aux Mois 3et6après l'instauration du traitement et au minimum chaque année pendant le traitement chronique.
Des augmentations de la glycémie ont été rapportées avec le traitement par nilotinib (voir rubrique Mises en garde spéciales et précautions d'emploi). La glycémie doit être évaluée avant l'instauration d'un traitement par nilotinib et surveillée pendant le traitement.
Population pédiatrique
La sécurité et l'efficacité du nilotinib chez les patients pédiatriques atteints de LMC chromosome Philadelphie positive en phase chronique âgés de 2 ans à moins de 18 ans ont été établies (voir rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques). On ne dispose d'aucune expérience chez les patients pédiatriques âgés de moins de 2 ans ou chez les patients pédiatriques atteints de LMC chromosome Philadelphie positive en phase accélérée ou en crise blastique. Il n'existe aucune donnée chez les patients pédiatriques nouvellement diagnostiqués âgés de moins de 10 ans et les données relatives aux patients pédiatriques résistants ou intolérants à l'imatinib âgés de moins de 6 ans sont limitées.
Mode d'administration
NILOTINIB BIOGARAN doit être pris deux fois par jour à 12 heures d'intervalle environ et ne doit pas être pris avec de la nourriture. Les gélules doivent être avalées entières avec de l'eau. Aucun aliment ne doit être consommé pendant les 2 heures précédant la prise du médicament et pendant une heure au moins après celle-ci.
Pour les patients présentant des difficultés à avaler, y compris les patients pédiatriques qui ne sont pas en mesure d'avaler des gélules, d'autres médicaments à base de nilotinib doivent être utilisés à la place de NILOTINIB BIOGARAN.
Durée de conservation :
2 ans
Précautions particulières de conservation :Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Des cas isolés de surdosage intentionnel avec le nilotinib ont été rapportés, dans lesquels le nombre de gélules de nilotinib ingérées avec de l'alcool et d'autres médicaments n'était pas précisé. Les événements ont inclus une neutropénie, des vomissements et un endormissement. Aucune modification de l'ECG ou hépatotoxicité n'ont été rapportées. Ces cas rapportés se sont résolus par un rétablissement.
En cas de surdosage, le patient doit être placé sous surveillance et recevoir un traitement de soutien approprié.
Classe pharmacothérapeutique : Antinéoplasiques, Inhibiteurs de la tyrosine kinase BCR-ABL, code ATC : L01EA03.
Mécanisme d'action
Le nilotinib est un inhibiteur puissant de l'activité tyrosine kinase ABL de l'oncoprotéine BCR-ABL, à la fois dans les lignées cellulaires et dans les cellules leucémiques primaires chromosome Philadelphie positives. La substance présente une forte affinité pour le site de liaison de l'ATP, ce qui en fait un inhibiteur puissant du BCR-ABL de type sauvage, également actif contre 32 des 33 formes mutantes du BCR-ABL résistantes à l'imatinib. En raison de cette activité biochimique, le nilotinib inhibe de manière sélective la prolifération et induit l'apoptose au niveau des lignées cellulaires et des cellules leucémiques primaires chromosome Philadelphie positives chez les patients atteints de LMC. Dans des modèles murins de LMC, le nilotinib en monothérapie réduit la charge tumorale et prolonge la survie après administration orale.
Effets pharmacodynamiques
Le nilotinib a peu ou pas d'effet contre la majorité des autres protéines kinases examinées (y compris Src), à l'exception des récepteurs des protéines kinases PDGF, KIT et Ephrine qu'il inhibe à des concentrations comprises dans l'intervalle atteint après une administration orale aux doses thérapeutiques recommandées dans le traitement de la LMC (voir tableau 4).
Tableau 4 Profil d'inhibition de l'activité kinase par le nilotinib (phosphorylation CI50 nM)
BCR-ABL | PDGFR | Kit |
20 | 69 | 210 |
Efficacité clinique
Etudes cliniques dans la LMC en phase chronique nouvellement diagnostiquée
Une étude clinique en ouvert, multicentrique, randomisée de phase III a été menée pour évaluer l'efficacité du nilotinib par rapport à l'imatinib chez 846 patients adultes atteints de LMC chromosome Philadelphie positive en phase chronique nouvellement diagnostiquée confirmée par analyse cytogénétique. Le diagnostic datait de moins de six mois et les patients n'étaient pas traités au préalable à l'exception des traitements par hydroxyurée et/ou anagrélide. Les patients ont été randomisés selon un rapport de 1/1/1 pour recevoir soit 300 mg de nilotinib deux fois par jour (n = 282), soit 400 mg de nilotinib deux fois par jour (n = 281), soit 400 mg d'imatinib une fois par jour (n = 283). La randomisation a été stratifiée en fonction du score de risque de Sokal au moment du diagnostic.
Les caractéristiques initiales étaient bien équilibrées dans les trois groupes de traitement. L'âge médian était de 47 ans dans les deux bras nilotinib et de 46 ans dans le bras imatinib. Les proportions de patients âgés de ≥ 65 ans étaient de 12,8 % dans le bras nilotinib à la dose de 300 mg deux fois par jour, 10,0 % dans le bras nilotinib à la dose de 400 mg deux fois par jour et 12,4 % dans le bras imatinib à la dose de 400 mg une fois par jour. Les trois groupes comptaient un peu plus d'hommes que de femmes (56,0 %, 62,3 % et 55,8 %, respectivement, dans les bras nilotinib à la dose de 300 mg deux fois par jour et à la dose de 400 mg deux fois par jour et dans le bras imatinib à la dose de 400 mg une fois par jour). Plus de 60 % des patients étaient caucasiens et 25 % des patients étaient asiatiques.
La date de première analyse était prévue lorsque tous les 846 patients avaient atteint 12 mois de traitement (ou arrêté prématurément le traitement). Les analyses ultérieures reflètent le moment où les patients avaient atteint 24, 36, 48, 60 et 72 mois de traitement (ou arrêté prématurément le traitement). La durée médiane de traitement a été d'environ 70 mois dans les groupes de traitement par nilotinib et 64 mois dans le groupe imatinib. La dose médiane réelle était de 593 mg/jour pour le nilotinib à la dose de 300 mg deux fois par jour, 772 mg/jour pour le nilotinib à la dose de 400 mg deux fois par jour et 400 mg/jour pour l'imatinib à la dose de 400 mg une fois par jour. Cette étude est en cours.
Le critère d'efficacité principal était la réponse moléculaire majeure (RMM) à 12 mois. La RMM était définie comme une valeur du ratio de BCR-ABL/ABL ≤ 0,1 % mesurée en RQ-PCR selon l'échelle internationale (EI), ce qui correspond à une réduction ≥ 3 log du transcrit BCR-ABL par rapport au taux initial standardisé. Le taux de RMM à 12 mois était significativement supérieur pour le nilotinib à la dose de 300 mg deux fois par jour par rapport à l'imatinib à la dose de 400 mg une fois par jour (44,3 % versus 22,3 %, p < 0,0001). Le taux de RMM à 12 mois était également significativement supérieur pour le nilotinib à la dose de 400 mg deux fois par jour par rapport à l'imatinib 400 mg une fois par jour (42,7 % versus 22,3 %, p < 0,0001).
Les taux de RMM à 3, 6, 9 et 12 mois étaient de 8,9 %, 33 %, 43,3 % et 44,3 % pour le nilotinib à la dose de 300 mg deux fois par jour, 5,0 %, 29,5 %, 38,1 % et 42,7 % pour le nilotinib à la dose de 400 mg deux fois par jour et 0,7 %, 12,0 %, 18,0 % et 22,3 % pour l'imatinib à la dose de 400 mg une fois par jour.
Les taux de RMM à 12, 24, 36, 48, 60 et 72 mois sont présentés dans le tableau 5.
Tableau 5 Taux de RMM
Nilotinib 300 mg deux fois par jour n = 282 (%) | Nilotinib 400 mg deux fois par jour n = 281 (%) | Imatinib 400 mg une fois par jour n = 283 (%) | |
RMM à 12 mois | |||
Réponse (IC à 95 %) | 44,31 (38,4 ; 50,3) | 42,71 (36,8 ; 48,7) | 22,3 (17,6 ; 27,6) |
RMM à 24 mois | |||
Réponse (IC à 95 %) | 61,71 (55,8 ; 67,4) | 59,11 (53,1 ; 64,9) | 37,5 (31,8 ; 43,4) |
RMM à 36 mois2 | |||
Réponse (IC à 95 %) | 58,51 (52,5 ; 64,3) | 57,31 (51,3 ; 63,2) | 38,5 (32,8 ; 44,5) |
RMM à 48 mois3 | |||
Réponse (IC à 95 %) | 59,91 (54,0 ; 65,7) | 55,2 (49,1 ; 61,1) | 43,8 (38,0 ; 49,8) |
RMM à 60 mois4 | |||
Réponse (IC à 95 %) | 62,8 (56,8 ; 68,4) | 61,2 (55,2 ; 66,9) | 49,1 (43,2 ; 55,1) |
RMM à 72 mois5 | |||
Réponse (IC à 95 %) | 52,5 (46,5 ; 58,4) | 57,7 (51,6 ; 63,5) | 41,7 (35,9 ; 47,7) |
1 Test de Cochran-Mantel-Haenszel (CMH), valeur de p pour le taux de réponse (versus imatinib 400 mg) < 0,0001
2 Seuls les patients présentant une RMM à un temps d'évaluation donné sont inclus comme répondeurs pour ce temps d'évaluation. Au total, 199 (35,2 %) patients n'étaient pas évaluables pour la RMM à 36 mois (87 dans le groupe nilotinib à la dose de 300 mg deux fois par jour et 112 dans le groupe imatinib) en raison d'évaluations de la PCR manquantes/non évaluables (n = 17), de la présence de transcrits atypiques à l'inclusion (n = 7) ou de l'arrêt de l'étude avant le Mois 36 (n = 175).
3 Seuls les patients présentant une RMM à un temps d'évaluation donné sont inclus comme répondeurs pour ce temps d'évaluation. Au total, 305 (36,1 %) patients n'étaient pas évaluables pour la RMM à 48 mois (98 dans le groupe nilotinib à la dose de 300 mg deux fois par jour, 88 dans le groupe nilotinib à la dose de 400 mg deux fois par jour et 119 dans le groupe imatinib) en raison d'évaluations de la PCR manquantes/non évaluables (n = 18), de la présence de transcrits atypiques à l'inclusion (n = 8) ou de l'arrêt de l'étude avant le temps d'évaluation Mois 48 (n = 279).
4 Seuls les patients présentant une RMM à un temps d'évaluation donné sont inclus comme répondeurs pour ce temps d'évaluation. Au total, 322 (38,1 %) patients n'étaient pas évaluables pour la RMM à 60 mois (99 dans le groupe nilotinib à la dose de 300 mg deux fois par jour, 93 dans le groupe nilotinib à la dose de 400 mg deux fois par jour et 130 dans le groupe imatinib) en raison d'évaluations de la PCR manquantes/non évaluables (n = 9), de la présence de transcrits atypiques à l'inclusion (n = 8) ou de l'arrêt de l'étude avant le temps d'évaluation Mois 60 (n = 305).
5 Seuls les patients présentant une RMM à un temps d'évaluation donné sont considérés comme répondeurs pour ce temps d'évaluation. Au total, 395 (46,7 %) patients n'étaient pas évaluables pour la RMM à 72 mois (130 dans le groupe nilotinib à la dose de 300 mg deux fois par jour, 110 dans le groupe nilotinib à la dose de 400 mg deux fois par jour et 155 dans le groupe imatinib) en raison d'évaluations de la PCR manquantes/non évaluables (n = 25), de la présence de transcrits atypiques à l'inclusion (n = 8) ou de l'arrêt de l'étude avant le temps d'évaluation Mois 72 (n = 362).
Les taux de RMM à différents temps d'évaluation (incluant les patients répondeurs ayant obtenu une RMM à ces temps d'évaluation ou avant comme répondeurs) sont présentés en incidence cumulée de RMM (voir figure 1).
Figure 1 Incidence cumulée de RMM
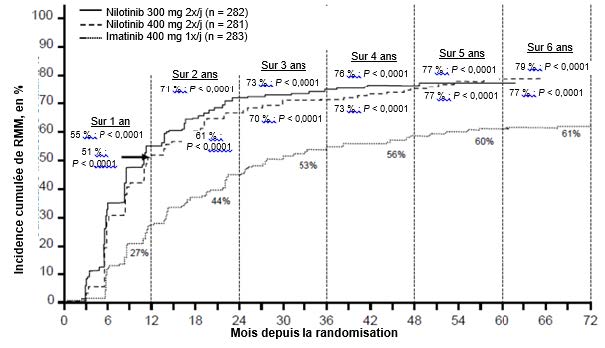
Pour tous les groupes de risque de Sokal, les taux de RMM à tous les temps d'évaluation sont restés systématiquement plus élevés dans les deux groupes nilotinib que dans le groupe imatinib.
Dans une analyse rétrospective, 91 % (234/258) des patients du bras nilotinib à la dose de 300 mg deux fois par jour ont présenté des taux de BCR-ABL ≤ 10 % après 3 mois de traitement contre 67 % (176/264) des patients du bras imatinib à la dose de 400 mg une fois par jour. Chez les patients présentant des taux de BCR ABL ≤ 10 % après 3 mois de traitement, la survie globale à 72 mois était supérieure à celle des patients ne présentant pas ce niveau de réponse moléculaire (94,5 % versus 77,1 % respectivement [p = 0,0005]).
Sur la base de l'analyse Kaplan-Meier du délai jusqu'à la première RMM, la probabilité d'atteindre une RMM à différents temps d'évaluation est plus élevée dans les deux bras nilotinib à la dose de 300 mg deux fois par jour et à la dose de 400 mg deux fois par jour que dans le bras imatinib à la dose de 400 mg une fois par jour, (RR = 2,17 et test du log-rank stratifié p < 0,0001 entre le nilotinib à la dose de 300 mg deux fois par jour et l'imatinib à la dose de 400 mg une fois par jour, RR = 1,88 et test du log-rank stratifié p < 0,0001 entre le nilotinib à la dose de 400 mg deux fois par jour et l'imatinib à la dose de 400 mg une fois par jour).
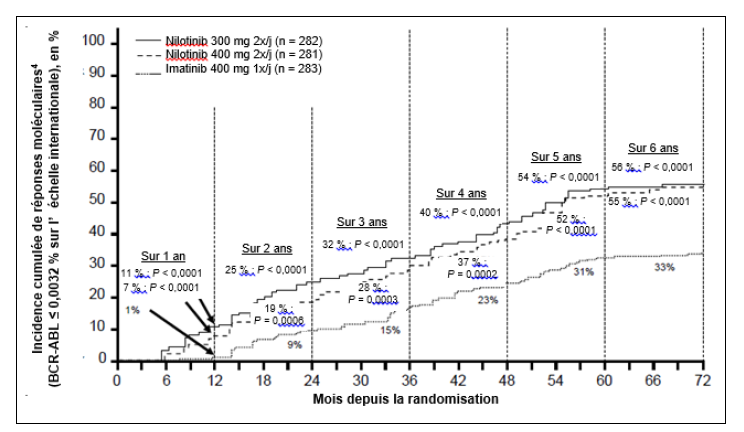
Les proportions de patients ayant obtenu une réponse moléculaire ≤ 0,01 % et ≤ 0,0032 % selon l'EI à différents temps d'évaluation sont présentées dans le tableau 6 et les proportions de patients ayant obtenu une réponse moléculaire ≤ 0,01 % et ≤ 0,0032 % selon l'EI aux différents temps d'évaluation sont présentées dans les figures 2 et 3. Des réponses moléculaires ≤ 0,01 % et ≤ 0,0032 % selon l'EI correspondent à une réduction respectivement ≥ 4 log et ≥ 4,5 log des transcrits BCR-ABL par rapport au taux initial standardisé.
Tableau 6 Proportions de patients ayant obtenu une réponse moléculaire ≤ 0,01 % (réduction de 4 log) et ≤ 0,0032 % (réduction de 4,5 log)
Nilotinib 300 mg deux fois par jour n = 282 (%) | Nilotinib 400 mg deux fois par jour n = 281 (%) | Imatinib 400 mg une fois par jour n = 283 (%) | ||||
= 0,01 % | = 0,0032 % | = 0,01 % | = 0,0032 % | = 0,01 % | = 0,0032 % | |
A 12 mois | 11,7 | 4,3 | 8,5 | 4,6 | 3,9 | 0,4 |
A 24 mois | 24,5 | 12,4 | 22,1 | 7,8 | 10,2 | 2,8 |
A 36 mois | 29,4 | 13,8 | 23,8 | 12,1 | 14,1 | 8,1 |
A 48 mois | 33,0 | 16,3 | 29,9 | 17,1 | 19,8 | 10,2 |
A 60 mois | 47,9 | 32,3 | 43,4 | 29,5 | 31,1 | 19,8 |
A 72 mois | 44,3 | 31,2 | 45,2 | 28,8 | 27,2 | 18,0 |
Figure 2 Incidence cumulée de réponses moléculaires ≤ 0,01 % (réduction de 4 log)
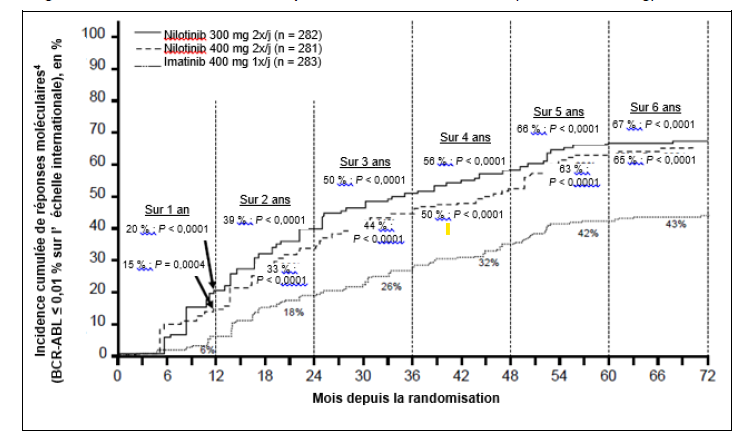
Figure 3 Incidence cumulée de réponses moléculaires ≤ 0,0032 % (réduction de 4,5 log)
Sur la base des estimations de Kaplan-Meier de la durée de première RMM, les proportions de patients ayant maintenu la réponse pendant 72 mois parmi ceux ayant obtenu une RMM étaient de 92,5 % (IC à 95 % : 88,6-96,4 %) dans le groupe nilotinib à la dose de 300 mg deux fois par jour, de 92,2 % (IC à 95 % : 88,5-95,9 %) dans le groupe nilotinib à la dose de 400 mg deux fois par jour et de 88,0 % (IC à 95 % : 83,0-93,1 %) dans le groupe imatinib à la dose de 400 mg une fois par jour.
La réponse cytogénétique complète (RCyC) était définie comme 0 % de métaphase Ph+ dans la moelle osseuse évaluée sur un minimum de 20 métaphases. Le meilleur taux de RCyC à 12 mois (les patients ayant obtenu une RCyC à 12 mois ou plus tôt étaient considérés comme répondeurs) a été significativement plus élevé dans les deux bras nilotinib à la dose de 300 mg et nilotinib à la dose de 400 mg deux fois par jour que dans le bras imatinib à la dose de 400 mg une fois par jour, voir tableau 7.
Le taux de RCyC sur 24 mois (incluant les patients ayant obtenu une RCyC à 24 mois ou plus tôt considérés comme répondeurs) a été statistiquement plus élevé dans les deux bras nilotinib à la dose de 300 mg et 400 mg deux fois par jour que dans le bras imatinib à la dose de 400 mg une fois par jour.
Tableau 7 Meilleur taux de RCyC
Nilotinib 300 mg deux fois par jour n = 282 (%) | Nilotinib 400 mg deux fois par jour n = 281 (%) | Imatinib 400 mg une fois par jour n = 283 (%) | |
Sur 12 mois | |||
Réponse (IC 95 %) | 80,1 (75,0 ; 84,6) | 77,9 (72,6 ; 82,6) | 65,0 (59,2 ; 70,6) |
Absence de réponse | 19,9 | 22,1 | 35,0 |
Valeur de p pour taux de réponse selon le test de CMH* (versus imatinib à la dose de 400 mg une fois par jour) | < 0,0001 | 0,0005 | |
Sur 24 mois | |||
Réponse (IC à 95 %) | 86,9 (82,4 ; 90,6) | 84,7 (79,9 ; 88,7) | 77,0 (71,7 ; 81,8) |
Absence de réponse | 13,1 | 15,3 | 23,0 |
Valeur de p pour taux de réponse selon le test de CMH* (versus imatinib à la dose de 400 mg une fois par jour) | 0,0018 | 0,0160 |
Sur la base des estimations de Kaplan-Meier, les proportions de patients ayant maintenu la réponse pendant 72 mois parmi les patients ayant obtenu une RCyC étaient de 99,1 % (IC à 95 % : 97,9-100 %) dans le groupe nilotinib à la dose de 300 mg deux fois par jour, de 98,7 % (IC à 95 % : 97,1-100 %) dans le groupe nilotinib à la dose de 400 mg deux fois par jour, et de 97,0 % (IC à 95 % : 94,7-99,4 %) dans le groupe imatinib à la dose de 400 mg une fois par jour.
La progression vers la phase accélérée (PA) ou la crise blastique (CB) sous traitement est définie comme le délai entre la date de randomisation et la première progression documentée en phase accélérée ou en crise blastique ou bien le décès lié à la LMC. La progression vers la phase accélérée ou la crise blastique sous traitement a été observée chez 17 patients au total : 2 patients traités par nilotinib à la dose de 300 mg deux fois par jour, 3 patients traités par nilotinib à la dose de 400 mg deux fois par jour et 12 patients traités par imatinib à la dose de 400 mg une fois par jour. Les pourcentages estimés de patients n'ayant pas progressé vers la phase accélérée ou la crise blastique à 72 mois ont été respectivement de 99,3 %, 98,7 % et 95,2 % (RR = 0,1599 et test du log-rank stratifié p = 0,0059 entre le nilotinib à la dose de 300 mg deux fois par jour et l'imatinib une fois par jour, RR = 0,2457 et test du log rank stratifié p = 0,0185 entre le nilotinib à la dose de 400 mg deux fois par jour et l'imatinib une fois par jour). Aucun nouveau cas de progression vers la PA/CB n'a été rapporté sous traitement depuis l'analyse à 2 ans.
En incluant l'évolution clonale comme critère de progression, 25 patients avaient progressé sous traitement vers la phase accélérée ou la crise blastique à la date d'analyse (3 dans le bras nilotinib à la dose de 300 mg deux fois par jour, 5 dans le bras nilotinib à la dose de 400 mg deux fois par jour et 17 dans le bras imatinib à la dose de 400 mg une fois par jour). Les taux estimés de patients n'ayant pas progressé vers la phase accélérée ou la crise blastique incluant l'évolution clonale à 72 mois ont été respectivement de 98,7 %, 97,9 % et 93,2 % (RR = 0,1626 et test du log-rank stratifié p = 0,0009 entre le nilotinib à la dose de 300 mg deux fois par jour et l'imatinib une fois par jour, RR = 0,2848 et test du log rank stratifié p = 0,0085 entre le nilotinib à la dose de 400 mg deux fois par jour et l'imatinib une fois par jour).
Au total, 55 patients sont décédés pendant le traitement ou pendant le suivi après l'arrêt du traitement (21 dans le groupe nilotinib à la dose de 300 mg deux fois par jour, 11 dans le groupe nilotinib à la dose de 400 mg deux fois par jour et 23 dans le groupe imatinib à la dose de 400 mg une fois par jour). Vingt-six (26) de ces 55 décès étaient liés à la LMC (6 dans le groupe nilotinib à la dose de 300 mg deux fois par jour, 4 dans le groupe nilotinib à la dose de 400 mg deux fois par jour et 16 dans le groupe imatinib à la dose de 400 mg une fois par jour). Les taux estimés de patients en vie à 72 mois étaient respectivement de 91,6 %, 95,8 % et 91,4 % (RR = 0,8934 et test du log-rank stratifié p = 0,7085 entre le nilotinib à la dose de 300 mg deux fois par jour et l'imatinib, RR = 0,4632 et test du log-rank stratifié p = 0,0314 entre le nilotinib à la dose de 400 mg deux fois par jour et l'imatinib). En ne prenant en compte que les événements constitués par les décès liés à la LMC, les taux estimés de survie globale à 72 mois étaient respectivement de 97,7 %, 98,5 % et 93,9 % (RR = 0,3694 et test du log-rank stratifié p = 0,0302 entre le nilotinib à la dose de 300 mg deux fois par jour et l'imatinib, RR = 0,2433 et test du log-rank stratifié p = 0,0061 entre le nilotinib à la dose de 400 mg deux fois par jour et l'imatinib).
Etudes cliniques dans la LMC en phase chronique et en phase accélérée en cas de résistance ou d'intolérance à l'imatinib
Une étude en ouvert de phase II, multicentrique et non contrôlée, a été menée afin de déterminer l'efficacité du nilotinib chez des patients adultes atteints de LMC et présentant une résistance ou une intolérance à l'imatinib ; les patients en phase chronique et les patients en phase accélérée ont été répartis dans des bras de traitement séparés. L'efficacité a été évaluée sur 321 patients en PC et 137 patients en PA inclus dans l'étude. La durée médiane de traitement était de 561 jours pour les patients en PC et de 264 jours pour les patients en PA (voir tableau 8).
Le nilotinib a été administré en continu (deux fois par jour, 2 heures après un repas et sans prise alimentaire pendant au moins une heure après l'administration), sauf en cas de signes évidents d'une réponse insuffisante ou d'une progression de la maladie. La dose était de 400 mg deux fois par jour et une augmentation de la posologie à 600 mg deux fois par jour était autorisée.
Tableau 8 Durée de l'exposition au nilotinib
Phase chronique n = 321 | Phase accélérée n = 137 | |
Durée médiane de traitement (jours) (25e-75e percentiles) | 561 (196-852) | 264 (115-595) |
La résistance à l'imatinib était définie comme l'absence d'une réponse hématologique complète (après 3 mois), d'une réponse cytogénétique (après 6 mois) ou d'une réponse cytogénétique majeure (après 12 mois) ou comme une progression de la maladie après une réponse cytogénétique ou hématologique antérieure. Les patients intolérants à l'imatinib étaient définis comme les patients ayant interrompu le traitement par imatinib en raison d'une toxicité et ne présentant aucune réponse cytogénétique majeure au moment de l'inclusion dans l'étude.
Au total, 73 % des patients étaient résistants à l'imatinib, tandis que 27 % étaient intolérants à l'imatinib. La majorité des patients avaient des antécédents de LMC, incluant un traitement antérieur intensif par d'autres agents antinéoplasiques, dont l'imatinib, l'hydroxyurée et l'interféron, et certains patients étaient même en échec d'une greffe de moelle osseuse (tableau 9). La dose maximale antérieure médiane d'imatinib avait été de 600 mg/jour. La dose maximale antérieure d'imatinib était = 600 mg/jour chez 74 % de tous les patients, avec 40 % des patients recevant des doses d'imatinib = 800 mg/jour.
Tableau 9 Caractéristiques des antécédents de LMC
Phase chronique (n = 321) | Phase accélérée (n = 137)* | |
Délai médian depuis le diagnostic, en mois | 58 | 71 |
(intervalle) | (5-275) | (2-298) |
Imatinib | ||
Patients résistants | 226 (70 %) | 109 (80 %) |
Patients intolérants sans RCyM | 95 (30 %) | 27 (20 %) |
Durée médiane du traitement par imatinib, en jours (25e-75e percentiles) | 975 (519-1 488) | 857 (424-1 497) |
Traitement antérieur par hydroxyurée | 83 % | 91 % |
Traitement antérieur par interféron | 58 % | 50 % |
Greffe de moelle osseuse antérieure | 7 % | 8 % |
* Information manquante sur le statut de résistance/intolérance à l'imatinib pour un patient. | ||
Chez les patients en PC, le critère d'évaluation principal était la réponse cytogénétique majeure (RCyM), définie comme l'élimination (réponse cytogénétique complète, RCyC) ou la réduction significative à < 35 % de métaphases Ph+ (réponse cytogénétique partielle) des cellules hématopoïétiques Ph+. Chez les patients en PC, la réponse hématologique complète (RHC) était un critère d'évaluation secondaire. Chez les patients en PA, le critère d'évaluation principal était la réponse hématologique (RH) globale confirmée, définie comme une réponse hématologique complète, l'absence de signes de leucémie ou le retour en phase chronique.
Phase chronique
Chez les 321 patients en PC, le taux de RCyM était de 51 %. Dans la plupart des cas, la RCyM survenait rapidement, dans les 3 mois (médiane : 2,8 mois) suivant le début du traitement par nilotinib et cette réponse se maintenait. Le délai médian d'obtention de la RCyC se situait juste au-delà de 3 mois (médiane : 3,4 mois).
Parmi les patients ayant obtenu une RCyM, 77 % (IC à 95 % : 70 %-84 %) maintenaient cette réponse à 24 mois. La durée médiane de la RCyM n'a pas été atteinte. Parmi les patients ayant obtenu une RCyC, 85 % (IC à 95 % : 78 %-93 %) maintenaient cette réponse à 24 mois. La durée médiane de la RCyC n'a pas été atteinte. Les patients ayant une RHC à l'inclusion obtenaient plus rapidement une RCyM (1,9 mois versus 2,8 mois). Parmi les patients en PC ne présentant aucune RHC à l'inclusion, 70 % ont obtenu une RHC et le délai médian d'obtention de cette RHC était de 1 mois ; la durée médiane de la RHC était de 32,8 mois. Le taux estimé de survie globale à 24 mois était de 87 % chez les patients atteints de LMC-PC.
Phase accélérée
Chez les 137 patients en PA, le taux de RH globale confirmée était de 50 %. Dans la plupart des cas, la RH survenait rapidement avec le traitement par nilotinib (médiane : 1,0 mois), et cette réponse était durable (la durée médiane de RH confirmée était de 24,2 mois). Parmi les patients ayant obtenu une RH, 53 % (IC à 95 % : 39 %-67 %) maintenaient cette réponse à 24 mois. Le taux de RCyM était de 30 % et le délai médian d'obtention de la réponse était de 2,8 mois. Parmi les patients ayant obtenu une RCyM, 63 % (IC à 95 % : 45 %-80 %) maintenaient cette réponse à 24 mois. La durée médiane de RCyM était de 32,7 mois. Le taux estimé de survie globale à 24 mois était de 70 % chez les patients atteints de LMC-PA.
Le tableau 10 présente les taux de réponse pour les deux bras de traitement.
Tableau 10 Réponse dans la LMC
(Meilleur taux de réponse) | Phase chronique | Phase accélérée | ||||
Intolérants (n = 95) | Résistants (n = 226) | Total (n = 321) | Intolérants (n = 27) | Résistants (n = 109) | Total* (n = 137) | |
Réponse hématologique (%) | ||||||
Globale (IC à 95 %) Complète ASL Retour en PC | - 87 (74-94) - - | - 65 (56-72) - - | - 701 (63-76) - | 48 (29-68) 37 7 4 | 51 (42-61) 28 10 13 | 50 (42-59) 30 9 11 |
Réponse cytogénétique (%) | ||||||
Majeure (IC à 95 %) Complète Partielle | 57 (46-67) 41 16 | 49 (42-56) 35 14 | 51 (46-57) 37 15 | 33 (17-54) 22 11 | 29 (21-39) 19 10 | 30 (22-38) 20 10 |
ASL = absence de signe de leucémie/réponse médullaire
1 114 patients en PC présentaient une RHC à l'état initial et n'étaient donc pas évaluables pour la réponse hématologique complète.
* Information manquante sur le statut de résistance/intolérance à l'imatinib pour un patient.
Les données d'efficacité chez les patients présentant une LMC-CB ne sont pas encore disponibles. Des bras de traitement séparés ont également été inclus dans l'étude de phase II, afin d'évaluer le nilotinib dans un groupe de patients en PC et en PA ayant reçu un traitement antérieur intensif par diverses thérapies, incluant un inhibiteur de la tyrosine kinase associé à l'imatinib. Parmi ces patients, 30/36 (83 %) étaient résistants mais non intolérants au traitement. Chez les 22 patients en PC évalués sur la base de l'efficacité, le nilotinib a induit un taux de RCyM de 32 % et un taux de RHC de 50 %. Chez les 11 patients en PA évalués sur la base de l'efficacité, le traitement a induit un taux de RH globale de 36 %.
Après l'échec du traitement par imatinib, 24 mutations BCR-ABL différentes ont été observées chez 42 % des patients en phase chronique et chez 54 % des patients en phase accélérée, évalués sur la base des mutations. Le nilotinib s'est révélé efficace chez les patients porteurs de diverses mutations du BCR-ABL associées à une résistance à l'imatinib, excepté pour la T315I.
Arrêt du traitement chez les patients adultes atteints de LMC Ph+ en phase chronique ayant été traités par nilotinib en traitement de première ligne et ayant obtenu le maintien d'une réponse moléculaire profonde
Au cours d'une étude en ouvert, à bras unique, 215 patients adultes atteints de LMC Ph+ en phase chronique traités par nilotinib en traitement de première ligne pendant = 2 ans et ayant atteint une RM4.5 telle que mesurée par le test de BCR-ABL MolecularMD MRDx, ont été recrutés pour poursuivre le traitement par nilotinib pendant 52 semaines supplémentaires (phase de consolidation au nilotinib). Sur ces 215 patients, 190 (88,4 %) sont entrés en phase de RST après avoir obtenu le maintien d'une réponse moléculaire profonde au cours de la phase de consolidation, définie par les critères suivants :
· les 4 dernières évaluations trimestrielles (effectuées toutes les 12 semaines) généraient un résultat de RM4.0 au moins (BCR-ABL/ABL ≤ 0,01 % selon l'EI), maintenu pendant un an
· la dernière évaluation générait un résultat de RM4.5 (BCR-ABL/ABL ≤ 0,0032 % selon l'EI)
· pas plus de deux évaluations générant un résultat situé entre RM4.0 et RM4.5 (0,0032 % selon l'EI < BCR-ABL/ABL ≤ 0,01 % selon l'EI).
Le critère d'évaluation principal était le pourcentage de patients présentant une RMM à 48 semaines après le début de la phase RST (en considérant tout patient nécessitant une reprise du traitement comme non-répondeur).
Tableau 11 Rémission sans traitement après un traitement de première ligne par nilotinib
Patients entrés dans la phase RST | 190 | |
semaines après le début de la phase RST | 48 semaines | 264 semaines |
patients présentant une RMM ou mieux | 98 (51,6 %, [IC à 95 % : 44,2 ; 58,9]) | 79[2] (41,6 %, IC à 95 % : 34,5 ; 48,9) |
Patients sortis de la phase RST | 93[1] | 109 |
en raison d'une perte de RMM | 88 (46,3 %) | 94 (49,5 %) |
pour d'autres raisons | 5 | 15 |
Patients ayant repris le traitement après la perte de RMM | 86 | 91 |
récupérant la RMM | 85 (98,8 %) | 90 (98,9 %) |
récupérant la RM4.5 | 76 (88,4 %) | 84 (92,3 %) |
[1] Un patient n'a pas présenté de perte de RMM à la Semaine 48 mais est sorti de la phase RST
[2] Pour 2 patients, l'évaluation de la PCR n'était pas disponible à la Semaine 264 ; par conséquent, leur réponse n'a pas été prise en compte lors de l'analyse du gel des données à la Semaine 264.
Le délai pour lequel 50 % des patients ayant repris le traitement ont récupéré la RMM et la RM4.5 était de 7 semaines et 12,9 semaines, respectivement. Le taux cumulé de RMM récupéré à 24 semaines après la reprise du traitement était de 97,8 % (89/91 patients) et de RM4.5 récupéré à 48 semaines de 91,2 % (83/91 patients).
La médiane de survie sans traitement (SST) estimée selon Kaplan-Meier était de 120,1 semaines (IC à 95 % : 36,9 ; non estimable [NE]) (figure 4) ; sur les 190 patients, 91 (47,9 %) n'avaient pas présenté d'événement de SST.
Figure 4 Estimation de Kaplan-Meier de la survie sans traitement après le début de la RST (population totale d'analyse)
Arrêt du traitement chez les patients adultes atteints de LMC en phase chronique ayant obtenu le maintien d'une réponse moléculaire profonde avec le traitement par nilotinib suite à un traitement préalable par l'imatinib
Au cours d'une étude en ouvert, à bras unique, 163 patients adultes atteints de LMC Ph+ en phase chronique recevant des inhibiteurs de la tyrosine kinase (ITK) pendant ≥ 3 ans (imatinib comme traitement initial par ITK pendant plus de 4 semaines sans RM4.5 documentée sous imatinib au moment du passage au nilotinib, puis passage au nilotinib pendant au moins deux ans) et qui ont obtenu une RM4.5 suite au traitement par nilotinib telle que mesurée par le test de BCR-ABL MolecularMD MRDx, ont été recrutés pour poursuivre le traitement par nilotinib pendant 52 semaines supplémentaires (phase de consolidation au nilotinib). Sur les 163 patients, 126 (77,3 %) sont entrés en phase RST après avoir obtenu le maintien d'une réponse moléculaire profonde au cours de la phase de consolidation, définie par le critère suivant :
· Les 4 dernières évaluations trimestrielles (effectuées toutes les 12 semaines) n'indiquaient aucune perte confirmée de RM4.5 (BCR-ABL/ABL ≤ 0,0032 % selon l'EI) pendant un an.
Le critère d'évaluation principal était la proportion de patients sans perte confirmée de RM4.0 ou perte de RMM dans les 48 semaines suivant l'arrêt du traitement.
Tableau 12 Rémission sans traitement après traitement par nilotinib suite à un traitement antérieur par imatinib
Patients entrés dans la phase RST | 126 | |
semaines après le début de la phase RST | 48 semaines | 264 semaines |
patients présentant une RMM, sans perte confirmée de RM4.0 et sans reprise du nilotinib | 73 (57,9 %, [IC à 95 % : 48,8 ; 66,7]) | 54 (42,9 % [54/126 ; IC à 95 % : 34,1 ; 52,0]) |
Patients sortis de la phase RST | 53 | 74[1] |
en raison d'une perte de RM4.0 ou de RMM | 53 (42,1 %) | 61 (82,4 %) |
pour d'autres raisons | 0 | 13 |
Patients ayant repris le traitement après la perte de RMM ou une perte confirmée de RM4.0 | 51 | 59 |
récupérant la RM4.0 | 48 (94,1 %) | 56 (94,9 %) |
récupérant la RM4.5 | 47 (92,2 %) | 54 (91,5 %) |
[1] Deux patients ont eu une RMM (évaluation de la PCR) à 264 semaines mais sont sortis ultérieurement sans nouvelle évaluation de la PCR.
Le délai médian estimé selon Kaplan-Meier pour récupérer la RM4.0 et la RM4.5 avec le nilotinib était de 11,1 semaines (IC à 95 % : 8,1 ; 12,1) et de 13,1 semaines (IC à 95 % : 12,0 ; 15,9), respectivement. Le taux cumulé de RM4.0 et RM4.5 récupéré à 48 semaines après la reprise du traitement était de 94,9 % (56/59 patients) et de 91,5 % (54/59 patients), respectivement.
La SST médiane estimée selon Kaplan-Meier est de 224 semaines (IC à 95 % : 39,9 ; NE) (figure 5) ; sur les 126 patients, 63 (50,0 %) n'avaient pas présenté d'événement de SST.
Figure 5 Estimation de Kaplan-Meier de la survie sans traitement après le début de la RST (population totale d'analyse)
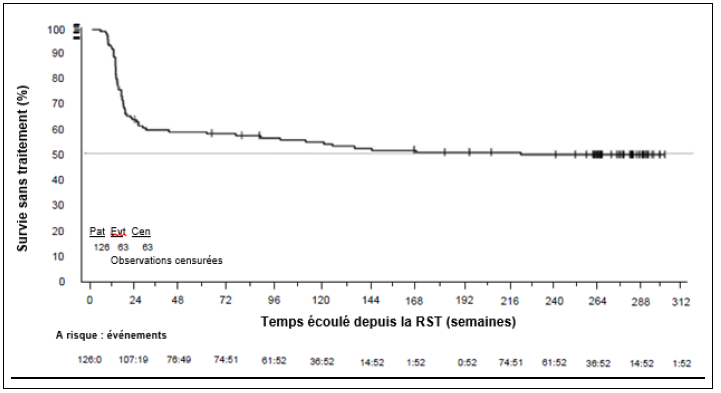
Population pédiatrique
Dans l'étude pédiatrique principale menée avec le nilotinib, 58 patients âgés de 2 à < 18 ans (25 patients nouvellement diagnostiqués avec une LMC Ph+ en phase chronique et 33 patients avec une LMC Ph+ en phase chronique résistants à l'imatinib/au dasatinib ou intolérants à l'imatinib) ont reçu un traitement par nilotinib à la dose de 230 mg/m2 deux fois par jour, arrondie aux 50 mg les plus proches (avec une dose maximale unique de 400 mg). Les principaux résultats de l'étude sont résumés dans le tableau 13.
Tableau 13 Résumé des données de l'étude pédiatrique principale menée avec le nilotinib
LMC-PC Ph+ nouvellement diagnostiquée (n = 25) | LMC-PC Ph+ résistante ou intolérante (n = 33) | |
Temps médian sous traitement en mois (intervalle) | 51,9 (1,4-61,2) | 60,5 (0,7-63,5) |
Intensité médiane (intervalle) de la dose effective (mg/m2/jour) | 377,0 (149-468) | 436,9 (196-493) |
Intensité relative de la dose (%) par rapport à la dose prévue de 230 mg/m2 deux fois par jour Médiane (intervalle) Nombre de patients avec > 90 % | 82,0 (32-102) 12 (48,0 %) | 95,0 (43-107) 19 (57,6 %) |
RMM (BCR-ABL/ABL = 0,1 % EI) à 12 cycles (IC à 95 %) | 60 % (38,7 ; 78,9) | 48,5 % (30,8 ; 66,5) |
RMM au Cycle 12 (IC à 95 %) | 64,0 %, (42,5 ; 82,0) | 57,6 %, (39,2 ; 74,5) |
RMM au Cycle 66 (IC à 95 %) | 76,0 %, (54,9 ; 90,6) | 60,6 %, (42,1 ; 77,1) |
Temps médian avant RMM en mois (IC à 95 %) | 5,56 (5,52 ; 10,84) | 2,79 (0,03 ; 5,75) |
Nombre de patients (%) atteignant la RM4.0 (BCR-ABL/ABL = 0,01 % EI) au Cycle 66 | 14 (56,0 %) | 9 (27,3 %) |
Nombre de patients (%) atteignant la RM4.5 (BCR-ABL/ABL = 0,0032 % EI) au Cycle 66 | 11 (44,0 %) | 4 (12,1 %) |
Perte de RMM confirmée parmi les patients ayant atteint la RMM | 3 sur 19 | Aucun sur 20 |
Mutation émergente sous traitement | Aucune | Aucune |
Progression de la maladie sous traitement | 1 patient a concordé temporairement à la définition technique de progression vers la PA/CB* | 1 patient a progressé vers la PA/CB après 10,1 mois sous traitement |
Survie globale Nbre d'événements Décès sous traitement Décès pendant le suivi de la survie | 0 3 (12 %) Non estimable | 0 1 (3 %) Non estimable |
* un patient a concordé temporairement avec la définition technique de progression vers la PA/CB (en raison d'une augmentation du nombre de basophiles), un mois après le début du traitement par nilotinib (avec interruption provisoire du traitement de 13 jours pendant le premier cycle). Le patient est resté dans l'étude, il est revenu en PC et était en RHC et en RCyC à 6 cycles de traitement par nilotinib.
Absorption
Les concentrations maximales du nilotinib sont atteintes 3 heures après une administration orale. Après administration orale, l'absorption du nilotinib est d'environ 30 %. La biodisponibilité absolue du nilotinib n'a pas été déterminée. Par rapport à une solution buvable (pH de 1,2 à 1,3), la biodisponibilité relative du nilotinib en gélule est d'environ 50 %. Chez des volontaires sains, en cas de prise alimentaire concomitante, la Cmax et l'aire sous la courbe concentration sérique-temps (ASC) du nilotinib augmentent respectivement de 112 % et 82 %, par rapport à une administration à jeun. L'administration de nilotinib 30 minutes ou 2 heures après la prise d'aliments augmente la biodisponibilité du nilotinib de 29 % et 15 %, respectivement (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
L'absorption du nilotinib (biodisponibilité relative) peut être réduite d'environ 48 % chez des patients ayant une gastrectomie totale et de 22 % chez des patients ayant une gastrectomie partielle, respectivement.
Distribution
Le rapport des concentrations sang/plasma du nilotinib est de 0,71. Le taux de liaison aux protéines plasmatiques est d'environ 98 %, sur la base des études in vitro.
Biotransformation
Les principales voies métaboliques identifiées chez les sujets sains sont l'oxydation et l'hydroxylation. Le nilotinib est le principal composant circulant dans le sérum. Aucun des métabolites ne contribue de manière significative à l'activité pharmacologique du nilotinib. Le nilotinib est principalement métabolisé par le CYP3A4, avec une éventuelle contribution mineure du CYP2C8.
Elimination
Après l'administration d'une dose unique de nilotinib marqué de manière radioactive chez des sujets sains, plus de 90 % de la dose étaient éliminés dans les 7 jours, principalement dans les selles (94 % de la dose). Le nilotinib inchangé représentait 69 % de la dose.
La demi-vie d'élimination apparente, estimée à partir de la pharmacocinétique à doses répétées avec une prise quotidienne, était d'environ 17 heures. La variabilité interindividuelle de la pharmacocinétique du nilotinib était modérée à élevée.
Linéarité/non-linéarité
A l'état d'équilibre, l'exposition au nilotinib dépendait de la dose. En cas d'une seule administration quotidienne de doses supérieures à 400 mg, les augmentations de l'exposition systémique étaient inférieures aux valeurs proportionnelles à la dose. En cas d'administration d'une dose de 400 mg deux fois par jour, l'exposition systémique quotidienne au nilotinib augmentait de 35 % à l'état d'équilibre, par rapport à l'exposition observée en cas d'une seule administration quotidienne d'une dose de 800 mg. L'exposition systémique (ASC) au nilotinib à l'état d'équilibre à une dose de 400 mg deux fois par jour était environ 13,4 % plus élevée qu'une dose de 300 mg deux fois par jour. La moyenne des valeurs hautes et basses de la concentration sur 12 mois était environ 15,7 % et 14,8 % plus élevée avec une dose de 400 mg deux fois par jour par rapport à une dose de 300 mg deux fois par jour. Aucune augmentation significative de l'exposition au nilotinib n'a été observée lors de l'augmentation de la posologie de 400 mg deux fois par jour à 600 mg deux fois par jour.
Les conditions d'équilibre sont généralement atteintes dans les 8 jours. Entre la prise de la première dose et l'état d'équilibre, l'exposition sérique au nilotinib augmentait d'environ 2 fois en cas d'une seule prise quotidienne, et d'environ 3,8 fois en cas de deux prises quotidiennes.
Pour les patients présentant des difficultés à avaler, y compris les patients pédiatriques qui ne sont pas en mesure d'avaler des gélules, d'autres médicaments à base de nilotinib doivent être utilisés à la place de NILOTINIB BIOGARAN.
Population pédiatrique
Après l'administration de nilotinib à des patients pédiatriques à la dose de 230 mg/m2 deux fois par jour, arrondie aux 50 mg les plus proches (jusqu'à une dose unique maximale de 400 mg), l'exposition à l'état d'équilibre et la clairance du nilotinib ont été similaires (dans les limites d'un facteur 2) par rapport aux patients adultes traités par 400 mg deux fois par jour. L'exposition pharmacocinétique du nilotinib après une ou plusieurs doses semblait comparable entre les patients pédiatriques âgés de 2 ans à < 10 ans et ceux âgés de ≥ 10 ans à < 18 ans.
Le nilotinib n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, il est recommandé aux patients présentant des sensations vertigineuses, une fatigue, des troubles de la vision ou d'autres effets indésirables susceptibles d'altérer leur aptitude à conduire des véhicules ou à utiliser des machines, de s'abstenir de mener ces activités tant que ces effets indésirables persistent (voir rubrique Effets indésirables).
Le nilotinib a été évalué dans des études de pharmacologie de sécurité, de toxicologie en administration répétée, de génotoxicité, de toxicité sur la reproduction, de phototoxicité et de cancérogenèse (chez le rat et la souris).
Etudes de pharmacologie de sécurité
Le nilotinib n'a exercé aucun effet sur le SNC ou les fonctions respiratoires. Les études de toxicité cardiaque in vitro ont révélé un signal préclinique d'allongement de l'intervalle QT, se traduisant par une inhibition des courants hERG et un allongement de la durée du potentiel d'action, induits par le nilotinib sur le coeur de lapins isolés. Chez des chiens ou des singes traités sur des périodes allant jusqu'à 39 semaines, et dans une étude spécifique de télémétrie réalisée chez le chien, aucun effet n'a été observé sur les paramètres de l'ECG.
Etudes de toxicologie en administration répétée
Des études de toxicologie en administration répétée jusqu'à 4 semaines chez le chien et jusqu'à 9 mois chez le singe cynomolgus, ont révélé que le foie était le principal organe cible de la toxicité du nilotinib.
Les altérations incluaient une élévation des activités de l'alanine aminotransférase et de la phosphatase alcaline, ainsi que des anomalies histopathologiques (essentiellement une hyperplasie/hypertrophie des cellules sinusoïdales ou des cellules de Kupffer, une hyperplasie des canaux biliaires et une fibrose périportale). En général, les modifications biochimiques étaient totalement réversibles après une période de récupération de quatre semaines, et les modifications histologiques étaient partiellement réversibles. Les expositions aux doses les plus faibles ayant induit des effets hépatiques étaient inférieures à l'exposition observée chez l'être humain traité à une dose de 800 mg/jour. Chez des souris et des rats traités pendant des durées allant jusqu'à 26 semaines, seuls des altérations hépatiques mineures ont été observées. Chez le rat, le chien et le singe, des augmentations le plus souvent réversibles des taux de cholestérol ont été observées.
Etudes de génotoxicité
Les études de génotoxicité menées sur des systèmes bactériens in vitro et des systèmes mammifères in vitro et in vivo, avec et sans activation métabolique, n'ont révélé aucun signe de potentiel mutagène du nilotinib.
Etudes de cancérogenèse
Dans l'étude de cancérogenèse de 2 ans chez le rat, l'utérus était le principal organe cible des lésions non néoplasiques (dilatation, ectasie vasculaire, hyperplasie des cellules endothéliales, inflammation et/ou hyperplasie épithéliale). Aucun signe de cancérogénicité n'a été mis en évidence suite à l'administration de nilotinib à 5, 15 et 40 mg/kg/jour. L'exposition (en termes d'ASC) à la dose la plus élevée a représenté environ 2 à 3 fois l'exposition journalière à l'état d'équilibre (sur la base de l'ASC) chez l'être humain recevant 800 mg/jour de nilotinib.
Dans l'étude Tg.rasH2 de cancérogenèse de 26 semaines chez la souris, dans laquelle le nilotinib a été administré à des doses de 30, 100 et 300 mg/kg/jour, des papillomes/carcinomes cutanés ont été détectés à la dose de 300 mg/kg, ce qui représente environ 30 à 40 fois (sur la base de l'ASC) l'exposition chez l'être humain à la dose maximale approuvée de 800 mg/jour (administrée à raison de 400 mg deux fois par jour). La dose n'induisant aucun effet de type lésions néoplasiques de la peau était de 100 mg/kg/jour, ce qui représente environ 10 à 20 fois l'exposition chez l'être humain à la dose maximale approuvée de 800 mg/jour (administrée à raison de 400 mg deux fois par jour). Les principaux organes cibles pour les lésions non néoplasiques étaient la peau (hyperplasie épidermique), les dents en croissance (dégénérescence/atrophie de l'émail des incisives supérieures et inflammation de la gencive/épithélium odontogène des incisives) et le thymus (augmentation de l'incidence et/ou de la sévérité de la diminution du nombre de lymphocytes).
Etudes de toxicité sur la reproduction et la fertilité
Le nilotinib n'induisait aucune tératogénicité, mais s'est révélé toxique pour l'embryon et le foetus à des doses entraînant également une toxicité maternelle. Lors de l'étude de fertilité réalisée chez les mâles et les femelles, ainsi qu'au cours de l'étude d'embryotoxicité uniquement réalisée chez les femelles, une augmentation des pertes post-implantation a été observée. Au cours des études d'embryotoxicité, une létalité embryonnaire et des effets sur le foetus (principalement une réduction du poids des foetus, une fusion prématurée des os de la face [fusion maxillaire/zygomatique] et des anomalies viscérales et squelettiques) chez le rat, ainsi qu'une augmentation de la résorption des foetus et des anomalies squelettiques chez le lapin, ont été observés. Dans une étude de développement pré- et post-natal menée chez le rat, l'exposition maternelle au nilotinib a entraîné une réduction du poids corporel associée à des modifications des paramètres du développement physique ainsi que des indices d'accouplement et de fertilité, chez la progéniture. Chez les femelles, l'exposition au nilotinib, aux doses n'induisant aucun effet indésirable, était généralement inférieure ou égale à l'exposition observée chez l'être humain à une dose de 800 mg/jour.
Aucun effet n'a été observé sur le nombre/la mobilité des spermatozoïdes ou sur la fertilité chez les rats mâles et femelles, traités jusqu'à la plus haute dose testée (environ 5 fois supérieure à la posologie recommandée chez l'être humain).
Etudes chez l'animal juvénile
Au cours d'une étude de développement menée chez l'animal juvénile, le nilotinib a été administré par voie orale à des rats juvéniles dès la première semaine post-partum jusqu'à l'âge adulte jeune (70 jours post-partum) à des doses de 2, 6 et 20 mg/kg/jour. Au-delà des paramètres standard d'étude, les évaluations des organes de référence pour le développement, les effets sur le SNC, sur l'accouplement et la fertilité ont été évalués. Sur la base d'une réduction du poids corporel dans les deux sexes et d'un retard de la séparation prépuciale chez les mâles (qui peut être associé à une perte de poids), la dose sans effet observable chez les rats juvéniles a été évaluée à 6 mg/kg/jour.
Les animaux juvéniles n'ont pas présenté d'augmentation de la sensibilité au nilotinib par rapport aux animaux adultes. De plus, le profil de toxicité des rats juvéniles a été comparable à celui observé chez les rats adultes.
Etudes de phototoxicité
Il a été démontré que le nilotinib absorbait la lumière dans la gamme UVB et UVA. Il a été distribué dans la peau et a présenté un potentiel phototoxique in vitro, mais aucun effet n'a été observé in vivo. Par conséquent, le risque de photosensibilisation par le nilotinib chez les patients est considéré comme très faible.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament soumis à une prescription initiale hospitalière semestrielle.
Prescription initiale et renouvellement réservés aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Remboursement en fonction de l'indication (JO du 29/10/2024) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- traitement des patients adultes atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique nouvellement diagnostiquée ;
- traitement des patients adultes atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique ou en phase accélérée, résistants ou intolérants à un traitement antérieur incluant l'imatinib ;
- traitement des patients pédiatriques atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique résistants ou intolérants à un traitement antérieur incluant l'imatinib.
Médicament soumis à une prescription initiale hospitalière semestrielle.
Prescription initiale et renouvellement réservés aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Remboursement en fonction de l'indication (JO du 29/10/2024) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- traitement des patients adultes atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique nouvellement diagnostiquée ;
- traitement des patients adultes atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique ou en phase accélérée, résistants ou intolérants à un traitement antérieur incluant l'imatinib ;
- traitement des patients pédiatriques atteints de leucémie myéloïde chronique (LMC) chromosome Philadelphie positive (Ph+) en phase chronique résistants ou intolérants à un traitement antérieur incluant l'imatinib.
Gélule.
Poudre blanche à jaunâtre dans une gélule en HPMC opaque de couleur jaune clair, de taille 0 (environ 21,4 mm de long), comportant la mention « 200 mg » imprimée en noir et à l'horizontale sur le corps.
Conditionnement unitaire contenant 28 × 1 gélules sous plaquettes prédécoupées unitaires en (PVC/PE/PVDC/Alu).
Nilotinib (sous forme de chlorhydrate dihydraté)..................................................................... 200 mg
Pour une gélule.
Excipient à effet notoire : chaque gélule contient 139 mg de lactose.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Lactose monohydraté, crospovidone de type A (E1202), silice colloïdale anhydre (E551), stéarate de magnésium (E470b).
Enveloppe de la gélule
Hypromellose (E464), carraghénanes (E407), chlorure de potassium (E508), oxyde de fer jaune (E172), dioxyde de titane (E171), eau purifiée.
Encre d'impression noire
Gomme laque (E904), propylèneglycol (E1520), hydroxyde de potassium (E525), oxyde de fer noir (E172).